Ryan Shienbaum, BS,
Gary Shienbaum, MD
Presentation
A 30-year-old Ukrainian male presented to Wills Eye Institute for the evaluation of progressive nyctalopia over the past three years. Additionally, the patient reported that he has had difficulty seeing moving objects over the past six months. He was no longer able to drive at night, as his visual responsiveness to an oncoming threat had become significantly impaired.
Medical History
The patient reported having no other medical problems. In the past, he was involved in amateur boxing, but denied any eye trauma during that time. Family history was unremarkable for ophthalmic disease. He denied tobacco and illicit drug use, and used alcohol socially.
Examination
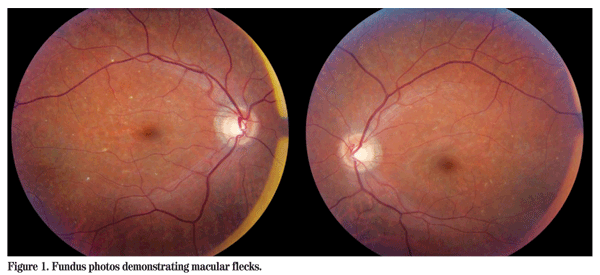
Visual acuity was 20/25 in the right eye and 20/30 in the left eye. Pupillary responses were normal and there was no relative afferent pupillary defect. There was no strabismus. Slit-lamp examination was unremarkable. Intraocular pressure by applanation tonometry was 10 mmHg OU. Ishihara color plates were test plate only in both eyes. Dilated fundus examination revealed a few scattered macular and midperipheral subretinal flecks in both eyes (See Figure 1). In the right eye there was some pigmentary disruption temporal to the macula, but the posterior pole was otherwise normal with the exception of a depleted nerve fiber layer reflex.
Diagnosis, Workup and Treatment
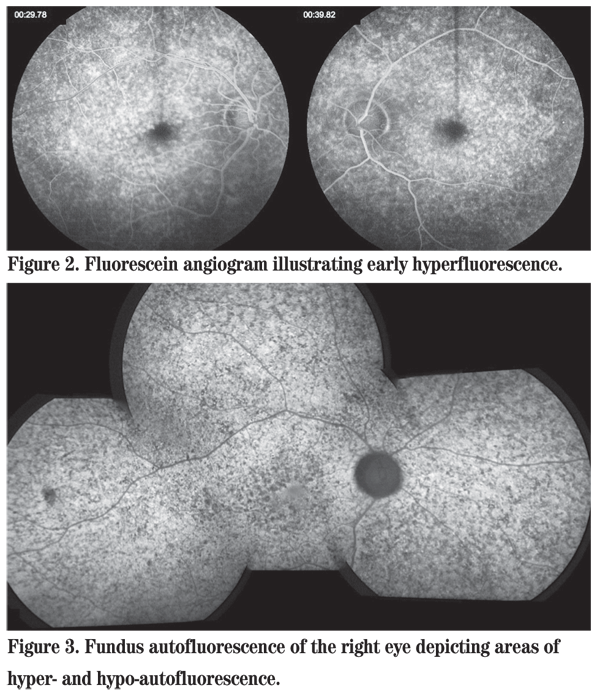
The differential diagnosis of an otherwise healthy young male presenting with nyctalopia and macular and midperipheral subretinal flecks is the family of fleck retinal dystrophies including Stargardt disease, fundus flavimaculatus, retinitis punctata albescens, familial fleck retina, and fleck retina of Kandori, among others. As the disorder in this patient is progressive, the stationary disorder fundus albipunctatus is ruled out. Fluorescein angiography revealed diffuse areas of early hyperfluorescence in both eyes (See Figure 2). Fundus autofluorescence demonstrated areas of hyperautofluorescence, representing an accumulation of lipofuscin, alongside areas of hypoautofluorescence, representing RPE atrophy or blockage, in both eyes (See Figure 3). An electroretinogram showed a bilateral reduction in amplitude and increased implicit time in response to both scotopic and photopic stimuli.
The patient had the opportunity to meet with an ocular genetics counselor, and an extended family pedigree revealed that his maternal and paternal family originated from small villages in the Ukraine less than 100 miles apart. This suggested a constricted gene pool, increasing the likelihood of an autosomal recessive retinal dystrophy. Subsequent genetic testing revealed two mutations within the patient's ABCA4 gene leading to the presumptive diagnosis of an ABCA4-related retinal dystrophy. The patient was found to be a compound heterozygote, as opposed to a homozygote as might be expected from a constricted gene pool. One mutation is a well-characterized pathogenic mutation; the other mutation is novel, but is predicted to cause abnormal splicing.
Discussion
The ABCA4 gene, which lies on the short arm of chromosome 1, encodes an ATP-binding cassette transporter that is expressed within retinal photoreceptors.1The encoded protein transports all-trans retinal from the photoreceptors to the retinal pigment epithelium. Mutations within the ABCA4 gene may result in functionally inactive transporters, causing all-trans retinal to accumulate in the photoreceptor outer segments. This results in N-retinyl-N-retinylidene ethanolamine (A2E) accumulation within lipofuscin storage granules in the RPE. Ultimately, A2E is cytotoxic to the RPE through mechanisms such as blue-light sensitization and resultant photochemical damage, complement activation and membrane destabilization.1-3
Despite the fact that most ABCA4-related dystrophies are autosomal recessive, and the most common phenotype is Stargardt disease, the ABCA4 gene has been implicated in several other phenotypically distinct disease processes, including fundus flavimaculatus, cone-rod dystrophy and retinitis pigmentosa.1,4 It has been suggested that the severity of the resultant phenotype relates to the remaining activity of the mutated gene product. Categorizing an ABCA4-related retinal dystrophy can be challenging, as its phenotypic manifestations often do not fit directly into a defined disease state. Instead, a spectrum exists, where a given ABCA4-related retinal dystrophy may lie along a continuum of phenotypes.4 This finding of multiple phenotypes derived from mutations in one gene is termed phenotypic heterogeneity.
Stargardt disease is classically described as an autosomal recessive, juvenile-onset macular dystrophy due to mutations in ABCA4 (STGD1) and represents the most common inherited macular dystrophy, having been estimated to affect one in every 10,000 people worldwide. Histo-pathologic analysis has revealed the accumulation of lipofuscin in the RPE, a feature common to the ABCA4-related retinal dystrophies.3,6 Additionally, there exist autosomal-dominant forms of Stargardt-like disease due to mutations within the ELOVL4 and PROM1 genes (STGD3 and STGD4, respectively).5,7
This highlights the concept of genetic heterogeneity, defined as one phenotype that can be caused by mutations in more than one gene.
Currently, no effective treatment options exist for patients with ABCA4-related retinal dystrophies. Some researchers have suggested that isotretinoin may prove to be beneficial as it acts to reduce levels of all-trans retinal.4,8 Similar reasoning has led some to speculate about a potential benefit to restricting excessive light exposure. This may, in theory, reduce photo-oxidative damage and decrease the formation of all-trans retinal from 11-cis retinal and, ultimately, the accumulation of A2E.4 Additionally, excessive vitamin A intake has been shown to result in an increased accumulation of lipofuscin within the RPE of an ABCA4 knockout mouse model. Consequently, vitamin A supplementation is discouraged in patients with ABCA4-related retinal dystrophies. Based on these studies vitamin A antagonists have been proposed as a potential treatment.9
This case illustrates that ABCA4 should be considered as the possible causative gene in flecked retinal dystrophies. Confirmation of a patient's genotype can lead to improved genetic counseling as well as modification of a treatment plan.
The authors thank Alex V. Levin MD, MHSc, and Eliza Stroh, MS, of the Pediatric Ophthalmology and Ocular Genetics Ser-vice at Wills Eye Institute for their assistance with this manuscript.
1. Molday RS, Zhong M, Quazi F. The role of the photoreceptor ABC transporter ABCA4 in lipid transport and Stargardt macular degeneration. Biochim Biophys Acta 2009;1791:573-583.
2. Walia S,
3. Kellner S, Kellner U, Weber BH, et al. Lipofuscin- and melanin-related fundus autofluorescence in patients with ABCA4- associated retinal dystrophies. Am J Ophthalmol 2009;147:895-902.
4. Klevering BJ, Deutman AF, Maugeri A, et al. The spectrum of retinal phenotypes caused by mutations in the ABCA4 gene. Graefe's Arch Clin Exp 2005;243:90-100.
5. Zhang K, Kniazeva M, Han M, et al. A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat Genet 2001;27:89-93.
6. Eagle RC, Lucier AC, Bernardino VB, Yanoff M. Retina pigment epithelial abnormalities in fundus flavimaculatus: A light and electron microscopic study. Ophthalmology 1980;87:1189-1200.
7. Kniazeva M, Chiang MF, Morgan B, et al. A new locus for autosomal dominant Stargardt-like disease maps to chromosome 4. Am J Hum Genet 1999;64:1394-1399.
8. Radu RA, Mata NL, Nusinowitz S, et al. Treatment with isotretinoin inhibits lipofuscin accumulation in a mouse model of recessive Star-gardt's macular degeneration. P Natl Acad Sci USA 2003;100:4742-4747.
9. Radu RA, Yuan Q, Hu J, et al. Accelerated accumulation of lipofuscin pigments in the RPE of a mouse model for ABCA4-mediated retinal dystrophies following vitamin A supplementation. Invest Ophth Vis Sci 2008;49:3821-3829.


