Stargardt disease was first described by German ophthalmologist Karl Stargardt in 1909 and remains the most common juvenile macular dystrophy, with an incidence of ~1/10,000 individuals worldwide. Clinically, patients typically present with central vision loss in childhood or early adulthood, central scotomata and irregular yellow-white outer retinal flecks, and an atrophic maculopathy which may be progressive throughout life. Choroidal neovascularization can rarely also develop in some cases. As our knowledge of the disorder has evolved, there’s been recognition of significant heterogeneity in the clinical findings, presentation, severity and course of STGD phenotypes among families with the disorder, even in patients with similar genotypes. Even though the previous term “Stargardt macular dystrophy” may describe the early posterior pole findings of the disease, in the past decade numerous studies have suggested it’s characterized by generalized retinal dysfunction as evidenced by ultra-widefield imaging and electrophysiologic testing.
Here, we’ll focus on the key aspects of Stargardt disease, from the pathophysiology to the most up-to-date diagnostic and treatment strategies in 2024.
Pathophysiology
The vast majority (>90 percent) of STGD cases are due to mutations in the ABCA4 gene, and are inherited in an autosomal-recessive manner. Additionally, Stargardt-like phenotypes may be inherited in an autosomal-dominant manner with mutations in PROM1 and ELOVL4.
The ABCA4 gene encodes the ATP-binding cassette (ABC) transporter protein in both rods and cones, which plays a crucial role in the transport of retinaldehyde across the retinal pigment epithelium and the photoreceptor cells. Dysfunction of the ABCA4 protein results in an accumulation of toxic products from the visual pathway within the photoreceptor outer segments which are then shed and phagocytized by the RPE, forming a major component of lipofuscin. The lipofuscin buildup leads to degeneration of the RPE and ultimately to the loss of photoreceptor cells.
One barrier to classifying and predicting STGD cases is there is substantial heterogeneity and over 800 mutations in ABCA4 autosomal recessive STGD with a wide range of effects on the protein function and expression. Therefore the phenotypes of ABCA4 STGD often exhibit large variability due to the underlying genetic heterogeneity. Additionally, certain types of mutations (truncating, severe misfolding, and intronic or splice variant mutations) may result in severe functional loss of ABC transport protein and severe disease.
Clinical Features and Diagnostic Testing
The clinical presentation will vary based on age of onset, presenting symptoms, fundus appearance and the severity of the underlying mutation, which can create some clinical uncertainty for a physician. As discussed above, the differences are most often explained by the severity of the mutations in the ABCA4 gene, the sensitivity of the foveal cones and RPE to these genomic differences.
The most common presenting complaint is usually a loss of central VA, which can range from 20/30 to 20/400. This loss of VA can occur as early as 5 years of age (i.e., the time of a potential failed vision screen) or in late adulthood (40 years or later). Usually the earlier and more severe conditions correlate to a severe ABCA4 genotype and more sensitive cones and RPE. Another reason why a patient presents to an ophthalmologist or retina specialist is due to an incidentally abnormal fundus exam during routine evaluation.
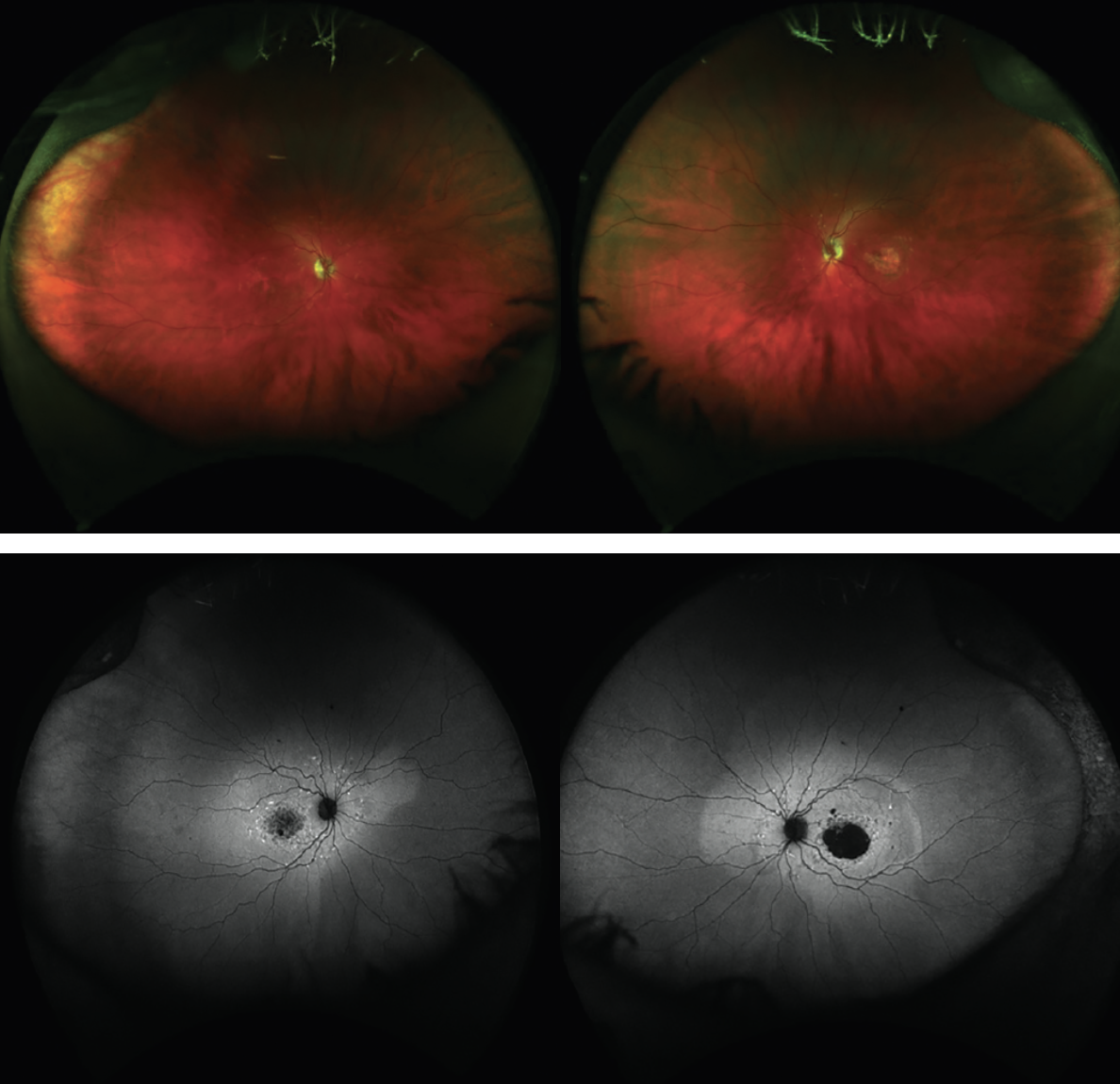 |
| Figure 1. Fundus photo and fundus autofluoresence of a young male with ABC4A (c2588GC)-confirmed Stargardt disease. |
The fundus exam often demonstrates lightly colored flecks at the level of the RPE. They may be differentiated from drusen as they’re usually more elongated, often in contact with each other and commonly extend beyond the macula. The flecks often meet, creating a fishtail appearance, hence the name pisciform flecks (Figure 1). Some patients will have a cluster of flecks two to three disc diameters around the fovea, while in others the flecks extend beyond the temporal vascular arcades up to or even beyond the equator. These flecks can also differ in several ways, but most notably in the amount and extent (pre-equatorial or extending anterior to the equator) to which they’re present. It’s possible some patients will exhibit minimal to no flecks, while others will have too many to count.
The fovea, on clinical exam, is often described as having a beaten-bronze appearance. This appearance is usually caused by the retention of bisretinoids (specifically A2E) intracellularly, resulting in a uniform light brown color that obscures choroidal details on ophthalmoscopy. This obscuration also contributes to the “dark or silent choroid” observed during fluorescein angiography, which is present in greater than 60 percent of patients (Figure 2). On FA, retinal blood vessels will hyperfluoresce against a hypofluorescent choroid. Flecks may appear hypofluorescent, or hyperfluorescent if associated with atrophy.
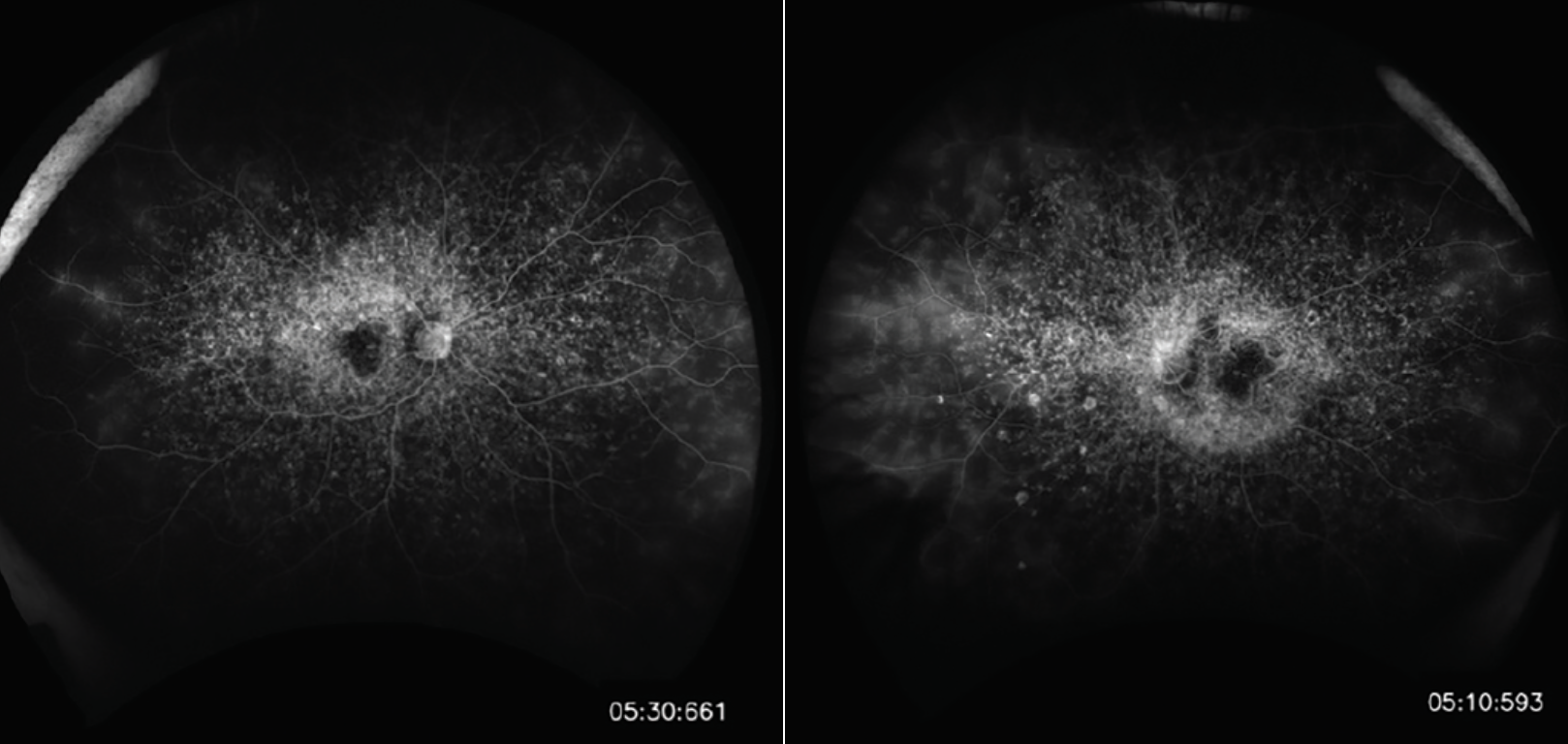 |
| Figure 2. Dark choroid on fluorescein angiography. |
Fluorescein angiography, along with ophthalmoscopy, will demonstrate relative sparing of the peripapillary RPE, which is diagnostic for Stargardt disease. Fundus autofluorescence is an excellent non-invasive alternative to FA and can further highlight flecks. Figure 3 demonstrates an SD-OCT image, which is a useful adjunct for illustrating the extent of outer retinal loss and RPE atrophy and can often detect early changes. The flecks appear as subretinal and intraretinal hyperreflective deposits. A sub-foveal optical gap, along with thickening of the external limiting membrane, hyperreflectivity at the base of the outer nuclear layer and disruption of the ellipsoid zone are also evident on OCT. Electrophysiology studies are typically normal or subnormal in full-field ERGs early in the disease stage but, over time, will show reduced responses secondary to photoreceptor degeneration. ERG can also be used to classify the severity and can be useful in tracking progression. Visual field testing may also accomplish a similar goal.
Though a choroidal neovascular membrane is a rare, late complication of STGD, it has the potential for severe visual impairment, if present. The CNVM can be treated with anti-VEGF if it occurs.
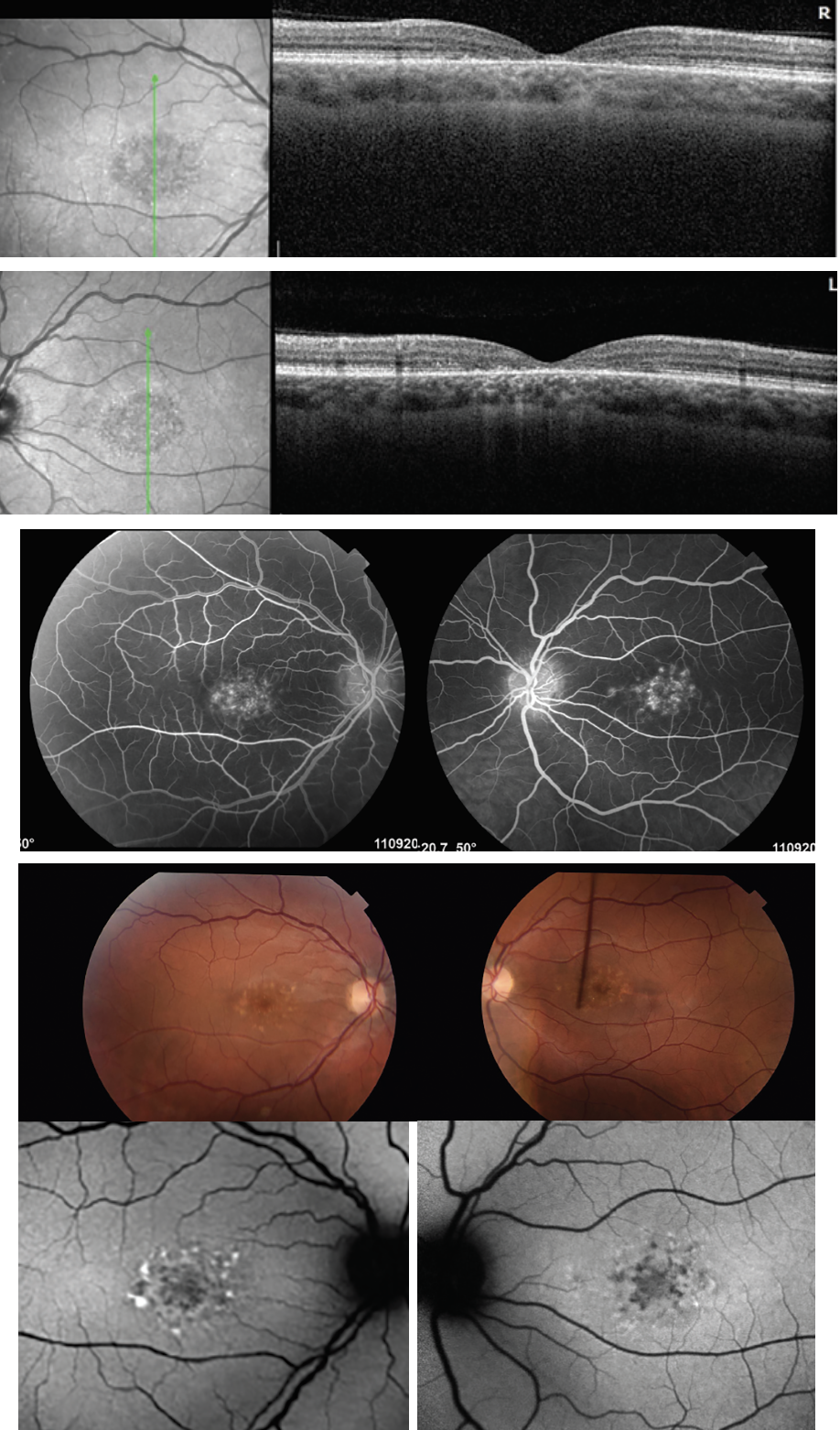 |
| Figure 3. Multimodal imaging, including OCT, demonstrating foveal thinning and central loss of RPE and outer retinal layer in ABC4A STGD. |
Genetic Testing
In cases of diagnostic uncertainty, and even in cases of confirmed STGD on clinical appearance, genetic testing should be performed, which offers several clinical benefits.
With inherited retinal degenerations in general, there can be significant phenotypic variability, and genetic testing can help conclusively determine the underlying molecular variant and provide more certainty to both physician and patient alike of the underlying mutation and clinical disease. Likewise, most clinical trials, as discussed below, will require genetic confirmation prior to entry into a trial. It’s important to involve a genetic counselor, because while most ABC4A STGD don’t have other non-ocular manifestations, there may be other relevant issues to address, such as prenatal counseling and family planning.
It’s important to remember that, in general, a causative mutation can be identified in up to 60 to 80 percent of patients with IRD. Often a saliva sample (2 mL) is sufficient for initial panel testing. For STGD, ABC4A mutations are relatively well-characterized. However, if panel testing doesn’t identify a variant, I’ll consider repeating the panel test in two to four years, as more loci and mutations are added yearly.
Furthermore, it bears mentioning that panel testing of a single individual doesn’t identify whether two mutations identified are in cis- or trans-configuration. Segregation analysis with a familial pedigree can often identify the inheritance pattern with greater precision, as well as if they’re on the same or different chromosomes. This can have important implications when a family is considering additional children or for their children’s reproductive future.
Management of Stargardt Disease
At this time, there are no proven treatments for Stargardt disease. Clinical trials may be an option depending on genetic testing results, age and risk/benefits of an investigational therapy (ongoing trials are discussed in greater detail below). Currently, there’s extensive research taking place in genetics, gene therapy and cell replacement therapy as possible therapeutic strategies. Given our knowledge of the underlying genetic derangement with the ABCA4 transmembrane protein, vitamin A is poorly processed and vitamin A byproducts are toxic. There’s some limited evidence that lower levels of vitamin A may be linked to a better visual outcome in STGD, but this also must be balanced against the need for vitamin A in other bodily functions, and patients should be counseled that vitamin A shouldn’t be completely cut out. Clinically, patients can be bothered by transitioning from one lighting environment to the next due to this particular dysfunction.
Other reasonable recommendations—though not well-studied—would be lutein supplementation (AREDS2 vitamins contain this), a diet rich with tomatoes (lycopene) and corn (zeaxanthin), as well as sunglasses/sun protection and avoidance of smoking. Lutein, lycopene and zeaxanthin accumulate on the inner surface of the retina and diminish the amount of blue light that’s transmitted to the photoreceptors. Involvement of a low-vision specialist or low-vision aids may be of use to patients for activities of daily living and should be proposed when necessary by a physician.
Clinical Trials And Potential Treatments
Given there are no current effective treatments for STGD, there’s great interest in cell, gene and pharmacologic therapies to potentially cure or slow this progressive disorder.
Cell therapy was initially explored in 2010, a Phase I/II safety and tolerability assessment for the use of hESC-derived RPE cells (named ASP7316 or MA09-hRPE) in STGD sponsored by Advanced Cell Technology, which has since been acquired by Astellas Pharma. In the U.S. trial arm, the subretinal delivery of stem cells via pars plana vitrectomy was safe and well-tolerated in 13 patients without failure or rejection. The most common postoperative complication was cataract progression following vitrectomy. Though visual acuity outcomes showed improvements in some patients, there was no control group. In 2024, the cell lines have undergone extension revision and Astellas plans ongoing trials in geographic atrophy secondary to age-related macular degeneration, and with hopes of expansion to STGD as well.
Pharmacologic intravitreal anti-complement agents that have gained FDA approval for geographic atrophy secondary to AMD have also completed trial enrollment and we eagerly await the results. A Phase IIB, randomized, double-masked, sham-controlled trial of the complement C5 inhibitor Zimura (Astellas) in subjects with autosomal recessive Stargardt disease has been fully enrolled and nears completion. The hypothesis is that reducing the inflammatory milieu with intravitreal complement inhibition may reduce the rate of atrophy progression as measured by en face OCT and fundus autofluorescence. Given good tolerability in the atrophic AMD population, we look forward to the results of this late-stage trial in the STGD population.
One downside of complement inhibition is that it doesn’t target the underlying cause of STGD, as a gene therapy would. However, there are limitations to gene therapy in ABC4A STGD given the size of the ABC4A gene. Well-established adenovirus vectors aren’t able to accommodate the size of the ABC4A gene, and therefore other viral vectors such as lentiviral vectors are under investigation. However, these other vectors require much preclinical and early clinical trial work.
In terms of gene therapy in the pipeline, Ocugen recently announced that dosing is complete in the third cohort of its Phase I/II GARDian clinical trial for OCU410ST (AAV-hRORA)—a modifier gene therapy candidate being developed for STGD. Three subjects received a single subretinal injection of the highest dose (2.25×1011 vg/mL) being tested.
Given the limitations of viral vectors, other methods, such as nanoparticles, offer an alternative transport method for larger transgenes. Another approach in early stages may be modulation of RNA via anti-sense oligonucleotides. Clustered regularly interspaced short palindrome repeat (CRISPR)-based technology is also under investigation for STGD as well as other IRDs.
Less-invasive approaches which may be favorable, especially in younger patients, are also under investigation and tend to focus on visual cycle inhibition to slow the progression and accumulation of toxic biproducts. For instance, emixustat hydrochloride from Acucela is being investigated in STGD. Emixustat is a potent visual cycle inhibitor of RPE65 isomerization activity and reduces visual chromophore (11-cis-retinal) production in a dose-dependent and reversible manner. One side effect of this type of therapy, however, is delayed dark adaptation due to the drug’s mechanism of action. Another oral visual cycle inhibitor, Tinlarebant (BeliteBio), is currently undergoing a Phase III clinical trial (DRAGON) in adolescent patients with Stargardt disease in hopes of slowing disease progression. Also, Nanoscope is currently developing a gene therapy that’s now in Phase II (STARLIGHT).
One other promising approach is oral administration of deuterated vitamin A (ALK-001/Gildeuretinol, Alkeus Pharmaceuticals). This drug has the C20 hydrogen atoms replaced with deuterium atoms, an isotope of hydrogen with a neutron in the nucleus; this impedes the dimerization of vitamin A and therefore reduces the opportunity for production of A2E. The hypothesis is that reducing levels of vitamin A will limit the production of all-trans-retinal and subsequently A2E. This compound is the subject of four clinical studies known as the TEASE trials, the initial results of which suggest good safety and tolerability and perhaps some encouraging early efficacy with regard to reducing the growth of atrophic lesions.
It’s important to note that, though several different therapeutic approaches have been discussed above, that list isn’t exhaustive. Other dietary supplementation approaches are being considered with various supplements, including docosahexaenoic acid (DHA), eicosapentaenoic acid (EPA) and alpha-linolenic acid (ALA) which are all omega-3 fatty acids and may support photoreceptor cells.
In conclusion, though Stargardt disease is one of the most common “juvenile macular dystrophies,” it remains a complex condition, and the search for therapeutic approaches is challenging. Genetic testing has become widely available in 2024, and is low cost, non-invasive and continues to expand our understanding, especially with the most common ABC4A STGD. Though there are currently no FDA-approved therapies or interventions for STGD, there are promising cell therapies, gene therapies and pharmacologic approaches, including intravitreal and oral methods, under investigation. At this time, once the diagnosis is established and genetic testing agrees, I tell patients there’s very likely to be some type of therapy in their lifetime. For now, supportive care with patient education, clinical trial referral when appropriate, low-vision services and monitoring by a retina or IRD specialist remain the mainstays of care.
Dr. Festok is a PGY-4 resident at Trinity Health Mid-Atlantic. Dr. Klufas practices at the Retina Service of Wills Eye Hospital in Philadelphia, and is an associate professor of ophthalmology at Thomas Jefferson University. Dr. Festok has no relevant financial interests. Dr. Klufas was principal ivestigator for the Zimura STGD Trial OPH2005 (IvericBio/Astellas). He can be reached at:
Thomas Jefferson University
840 Walnut Street, Suite 1020
Philadelphia, PA 19107
Tel: (800) 331-6634
www.midatlanticretina.com
www.willseye.org
1. Allikmets R, Singh N, Sun H, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet 1997;15:3:236-246.
2. Al-Khuzaei S, Broadgate S, Foster CR, et al. An overview of the genetics of ABCA4 retinopathies, an evolving story. Genes (Basel) 2021;12:8:1241.
3. Colella P, Auricchio A. AAV-mediated gene supply for treatment of degenerative and neovascular retinal diseases. Curr Gene Ther 2010;10:5:371-380.
4. Fujinami K, Lois N, Davidson AE, et al. A longitudinal study of Stargardt disease: Clinical and electrophysiologic assessment, progression, and genotype correlations. Am J Ophthalmol 2013;155:6:1075-1088.e13.
5. Georgiou M, Kane T, Tanna P, et al. Prospective cohort study of childhood-onset Stargardt disease: Fundus autofluorescence imaging, progression, comparison with adult-onset disease, and disease symmetry. Am J Ophthalmol 2020;211:159-175.
6. Huang D, Heath Jeffery RC, Aung-Htut MT, McLenachan S, Fletcher S, Wilton SD, Chen FK. Stargardt disease and progress in therapeutic strategies. Ophthalmic Genet 2021;43:1:1-26.
7. Klufas MA, Tsui I, Sadda SR, et al. Ultrawidefield autofluorescence in ABCA4 Stargardt disease. Retina 2018;38:2:403-415.
8. Kohli P, Tripathy K, Kaur K. Stargardt disease. [Updated 2024 Jan 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan. Available from: https://www.ncbi.nlm.nih.gov/books/NBK587351/.
9. Parker MA, Erker LR, Audo I, et al. Three-year safety results of SAR422459 (EIAV-ABCA4) gene therapy in patients with ABCA4-associated Stargardt disease: An open-label dose-escalation phase I/IIa clinical trial, cohorts 1-5. Am J Ophthalmol. 2022;240:285-301.
10. Piotter E, McClements ME, MacLaren RE. Therapy approaches for Stargardt disease. Biomolecules 2021;11:8:1179.
11. Schwartz SD, Regillo CD, Lam BL, et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: Follow-up of two open-label phase 1/2 studies. Lancet 2015;385:9967:509-516.
12. Tanna P, Strauss RW, Fujinami K, et al. Stargardt disease: Clinical features, molecular genetics, animal models, and therapeutic options. Br J Ophthalmol 2017;101:1:25-30.
13. University of Iowa Department of Ophthalmology and Visual Sciences. Stargardt disease. Accessed August 19, 2024. https://webeye.ophth.uiowa.edu/eyeforum/atlas/pages/Stargardt-disease-13.htm#gsc.tab=0
14. Sofi F, Sodi A, Franco F, et al. Dietary profile of patients with Stargardt’s disease and retinitis pigmentosa: Is there a role for a nutritional approach? BMC Ophthalmol 2016;16:13.
15. Stargardt K. Über familiare, progressive degeneration in der maculagegend des auges [article in German]. Albrecht von Graefes Arch Klin Exp Ophthalmol 1909;71:534-550.


