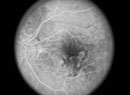
Late stage age-related macular degeneration is classified as dry or wet according to characteristic features of these two pathogenic modes. The dry form, which in its end stage is termed geographic atrophy, is characterized by localized regions of atrophy of the retinal pigment epithelium with associated overlying regions of photoreceptor cell death. The wet form, also called neovascular AMD, involves the growth of new blood vessels from the choroid capillary network across the Bruch's membrane/RPE interface into the neural retina, which results in retinal detachment, subretinal and intraretinal edema, and scarring.
While these late stages show different pathologies, early stages of AMD show similarities in pathogenesis. RPE cells form the outer blood-retinal barrier and provide essential photoreceptor support functions. Dysfunction in the RPE layer is clinically manifested as altered pigmentation and lipofuscin accumulation within the macular region. Drusen accumulation between the RPE and Bruch's membrane and alterations in structural and biochemical properties of Bruch's membrane are similarly associated with aging and early-stage AMD. However, events responsible for the initial insult to the retina and progression to late-stage AMD are not currently understood.1,2
Within the last few years, there have been some exciting advancements in treating the wet form of AMD, including verteporfin photodynamic therapy and development of anti-vascular endothelial growth factor (VEGF) compounds, such as pegaptanib sodium (Macugen, OSI/Eyetech) and ranibizumab (Lucentis, Genentech).3,4 Effective treatment options for dry AMD are not currently available, however.
AMD has been classified as a complex disease with multiple genetic and environmental risk factors. Risk factors for AMD include increased sunlight exposure, Caucasian race, female gender, increased dietary fat and smoking. Mutations in ApoE, ABCA4, FBLN5 and HEMICENTIN-1 genes have been reported to contribute, in part, to AMD progression.5-12 Early evidence of a genetic component for AMD was reported in a classical study showing hard and soft drusen formation correlated with 57 percent increased AMD risk in monozygotic (identical) twins and 81 percent increased risk in dizygotic (maternal) twins. However, until recently, genetic factors predisposing a large population of patients to AMD and pointing to a central mechanistic pathway in the pathophysiology of this disease have remained obscure. More recent genetic studies are closing this gap.
AMD Pathology
|
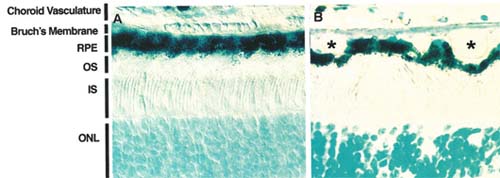
Molecular properties of the choroid/ Bruch's membrane/RPE axis and subsequent perturbations occurring during aging have been extensively studied in order to identify the mechanistic pathways responsible for AMD pathogenesis. Alterations in pigmentation, accumulation of toxic lipofuscin pigments, and formation of drusen, which are extracellular deposits that accumulate between the RPE and Bruch's membrane, are all hallmark features of AMD (See Figure 1).
The RPE plays a critical role in the support of photoreceptor cells. In addition to providing a selective cellular filtration component to the outer blood-retinal barrier, the RPE directly aids the photoreceptor by enzymatic re-isomerization of the visual chromophore and ensuring the daily turnover of photoreceptor shed disc membranes.13 These roles have been verified by identification of mutations in RPE genes affecting these functions, which result in photoreceptor degeneration. Lipofuscin in the RPE is a byproduct of incomplete outer segment digestion. These highly autofluorescent retinoids accumulate (See Figure 2) when the aging RPE is unable to completely digest the disc membranes.
|
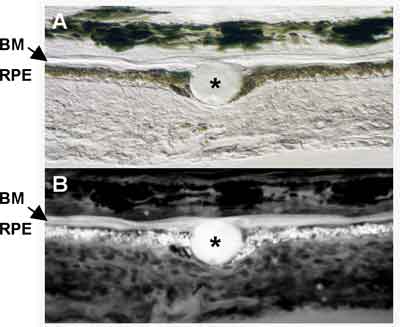
Drusen, composed of cellular debris and modified proteins and lipids from the RPE and Bruch's membrane, are associated with aging and believed to predispose the retina to disease progression. Drusen are subcategorized into both hard and soft forms, with the soft form conferring a significantly higher risk for AMD progression. Studies show compositional similarities between drusen and amyloid deposits, atherosclerotic plaques and dense deposits in atherosclerosis, amyloidosis, elastosis, and dense deposit disease associated with membranoproliferative glomerulonephritis type II.16-18 Studies show that molecular and cellular components of drusen are consistent with local cellular (RPE) dysfunction and atrophy.
Targeting Lipofuscin, A2E Formation
Accelerated lipofuscin and A2E accumulation occurs in patients with AMD and Stargardt macular dystrophy, a juvenile form of macular degeneration sharing many pathological features of AMD, and in mouse models of these retinal disorders. Accumulation of A2E has been associated with RPE dysfunction and atrophy and photoreceptor loss. As such, recent efforts have centered on targeting A2E formation in RPE cells as a therapeutic strategy for AMD and AMD-like retinal disorders. In early studies, it was shown that reducing dietary levels of vitamin A (all-trans-retinol) inhibits lipofuscin and autofluorescence accumulation in the RPE.19 While impractical as a therapy, these results led to developing an alternative approach for inhibiting vitamin A delivery specifically to the retina.
|
An alternative strategy for reducing retinol levels specifically in the retina was examined.20 N-(4-hydroxyphenyl) retinamide (HPR) (fenretinide) is a synthetic amide of all-trans-retinoic acid that has been used for decades, most commonly in cancer treatments, to remove serum retinol bound to carrier protein, retinol-binding protein (RBP).20 While this treatment has relatively few side effects, treated patients reported effects on night vision. It was subsequently shown that while additional retinol-binding proteins can compensate for retinol delivery to other tissues, the eye relies primarily on RBP for its retinol supply.21 Therefore, the selective effect of HPR treatments on retinol delivery to the eye was the basis for testing the efficacy of this drug on A2E formation in the ABCA4-/- Stargardt mouse model. HPR treaents inhibited the formation of RPE lipofuscin and A2E in these mice. However, prolonged treatment studies are needed, and efficacy in inhibiting the degeneration of RPE and photoreceptors has not yet been ascertained. More potent inhibitors of the retinoid (visual) cycle with prolonged drug activities have been identified.22 These compounds will need to be further developed before human trials can begin.
Alternatively, death of photoreceptor and RPE cells could result from the accumulation of visual pigments uncoupled from the chromophore.23 Thus, supplementation of the chromophore for visual pigment regeneration might be required.24 These two individual strategies, or the combination of both, will require direct testing in humans to determine optimal results.
A New Therapeutic Target
When RPE cells die, debris serves as a "nucleation" site for drusen formation and subsequent tissue inflammatory responses.25 Proteomic analysis of drusen from normal vs. AMD patients shows evidence of oxidative protein modifications that may contribute to drusen formation and AMD pathogenesis.26 Drusen formation in the aging eye has been compared to other aging disorders, such as Alzheimer's disease and atherosclerosis, where plaque formation and deposits induce chronic inflammatory stimuli and contribute to disease progression. Complement C3-, C5- and C5-9 activation components have also been identified in drusen.27,28
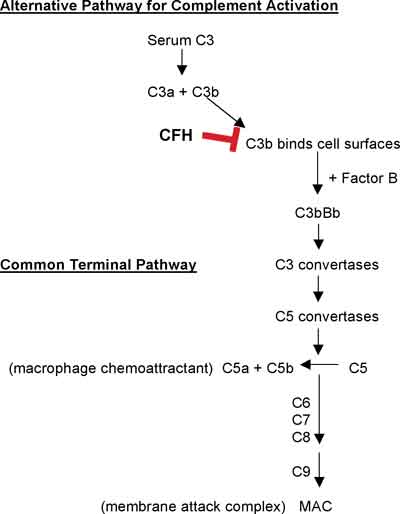
The alternative complement pathway is initiated by the spontaneous hydrolysis of plasma C3 to C3a and C3b (See Figure 3).29 Unregulated C3b binds to cell surfaces, thereby providing a mechanism for complement activation that will proceed unless downregulated by complement regulating proteins. This mechanism is the basis for the alternative pathway of complement activation. Complement factor H (CFH), an essential member of this regulatory pathway, is a plasma glycoprotein that binds to cell surface sialic acids or neutral or anionic polysaccharides, as well as to C3b. This interaction accelerates C3b degradation and inhibits C3 convertase activation. C3b not neutralized by CFH is available for binding to factor B, resulting in activation of the alternative pathway. In brief, C3b stabilization results in C3-convertase activation, converting C3 to C3a and C3b, with subsequent convertase activation to produce C5a (a macrophage chemoattractant) and C5b. C5b, combines with C6, C7, C8, and C9 to form a membrane attack complex (MAC), with C5b-9 in the terminal complement pathway. Therefore, production of C5a and C5b-9 activates, promotes and amplifies inflammatory responses.
Complement System and AMD
Considerable advances have been made in the last year linking the complement system to AMD. Several laboratories reported a significant association between AMD risk and mutations in the gene for CFH, the important regulator of the alternative complement pathway (See Figure 3).30-33 A tyrosine residue at position 402 replaced by a histidine residue (a T to C substitution at nucleotide 1277 in exon 9) shows allele frequency between 0.61 to 0.94 in AMD cases and 0.34 to 0.46 in age-matched controls (collectively) among these studies. Further, this variant confers a strong risk for soft drusen formation and both dry and wet forms of AMD.34
In a recent study, haplotype analysis of patients carrying the Y402H variant who do not develop AMD revealed factor B and complement component 2 gene variants that confer a protective effect, seemingly counterbalancing effects from the CFH variant.35 These results strongly support a central role for immune system regulation in the pathogenesis of AMD.
In support of these results, Ccl-2 (monocyte chemoattractant protein, MCP-1) and Ccr-2 (MCP-1 receptor) knockout mice develop AMD-like pathology including drusen formation, thickening of Bruch's membrane, lipofuscin and A2E accumulation in RPE cells, and photoreceptor degeneration. Normally, choroidal macrophages are attracted to C5a and IgG deposits in drusen for subsequent degradation of these immune complexes. Therefore, loss of MCP-1 and its receptor by genetic ablation in this mouse model was proposed to impair the recruitment of macrophages, resulting in chronic complement activation and tissue pathogenesis in these mice.36
The recent genetic evidence correlating complement factors with AMD risk strongly supports a major inflammatory component for AMD pathogenesis. While previous identification of environmental and genetic risk factors have contributed to our knowledge of retinal aging disorders, recent identification of immune system involvement fills an important gap in our understanding of the pathogenesis of AMD. In addition, the current research to control the formation of photosensitizing fluorophores such as A2E during aging represents an equally exciting approach for future therapies. Future research will focus on these pathogenic mechanisms in the aging eye. In addition, new strategies can now be developed that specifically address these injurious pathways, and may finally provide a therapeutic option for dry AMD.
Dr. Howes and Dr. Baehr are at the Moran Eye Center at the University of Utah, and Dr. Palczewski is in the Department of Pharmacology, Case School of Medicine, Case Western Reserve University, Cleveland. Contact Dr. Howes at the Department of Ophthalmology and Visual Sciences, University of Utah Health Science Center, Salt Lake City, Utah 84112. Phone (801) 585-0499; E-mail:
kim.howes@hsc.utah.edu.
1. Klein R, Peto T, Bird A, Vannewkirk MR. The epidemiology of age-related macular degeneration. Am J Ophthalmol 2004;137:486-95.
2. Johnson PT, Lewis GP, Talaga KC, et al. Drusen-associated degeneration in the retina. Invest Ophthalmol Vis Sci 2003;44:4481-8.
3. D'Amico DJ. Pegaptanib Sodium for Neovascular Age-Related Macular Degeneration: Two-Year Safety Results of the Two Prospective, Multicenter, Controlled Clinical Trials. Ophthalmology 2006; 113:1001.e1-6. Epub 2006 Apr 27.
4. Spaide RF, Sorenson J, Maranan L. Combined photodynamic therapy with verteporfin and intravitreal triamcinolone acetonide for choroidal neovascularization. Ophthalmology 2003;110:1517-25.
5. Klaver CC, Kliffen M, van Duijn CM, et al. Genetic association of apolipoprotein E with age-related macular degeneration. Am J Hum Genet 1998;63: 200-6.
6. Souied EH, Benlian P, Amouyel P, et al. The epsilon4 allele of the apolipoprotein E gene as a potential protective factor for exudative age-related macular degeneration. Am J Ophthalmol 1998;125:353-9.
7. Allikmets R, Shroyer NF, Singh N, et al. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science 1997;277:1805-7.
8. Allikmets R, Singh N, Sun H, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet 1997;15:236-46.
9. Stone EM, Braun TA, Russell SR, et al. Missense variations in the fibulin 5 gene and age-related macular degeneration. N Engl J Med 2004;351:346-53.
10. Schultz DW, Klein ML, Humpert AJ, et al. Analysis of the ARMD1 locus: Evidence that a mutation in HEMICENTIN-1 is associated with age-related macular degeneration in a large family. Hum Mol Genet 2003;12:3315-23.
11. Malek G, Johnson LV, Mace BE, et al. Apolipoprotein E allele-dependent pathogenesis: a model for age-related retinal degeneration. Proc Natl Acad Sci U S A 2005;102:11900-5.
12. Baird PN, Richardson AJ, Robman LD, et al. Apolipoprotein (APOE) gene is associated with progression of age-related macular degeneration (AMD). Hum Mutat 2006;27:337-42.
13. McBee JK, Palczewski K, Baehr W, Pepperberg DR. Confronting complexity: The interlink of phototransduction and retinoid metabolism in the vertebrate retina. Prog Retin Eye Res 2001;20:469-529.
14. Radu RA, Mata NL, Bagla A, Travis GH. Light exposure stimulates formation of A2E oxiranes in a mouse model of Stargardt's macular degeneration. PNAS 2004;101:5928-33.
15. De S, Sakmar TP. Interaction of A2E with Model Membranes. Implications to the Pathogenesis of Age-related Macular Degeneration. J. Gen. Physiol. 2002;120:147-57.
16. Mullins RF, Russell SR, Anderson DH, Hageman GS. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. Faseb J 2000;14:835-46.
17. Luibl V, Isas JM, Kayed R, et al. Drusen deposits associated with aging and age-related macular degeneration contain nonfibrillar amyloid oligomers. J Clin Invest 2006;116:378-85.
18. Yoshida T, Ohno-Matsui K, Ichinose S, et al. The potential role of amyloid beta in the pathogenesis of age-related macular degeneration. J Clin Invest 2005;115:2793-800.
19. Katz ML, Norberg M. Influence of dietary vitamin A on autofluorescence of leupeptin-induced inclusions in the retinal pigment epithelium. Exp Eye Res 1992;54:239-46.
20. Radu RA, Han Y, Bui TV, et al. Reductions in serum vitamin A arrest accumulation of toxic retinal fluorophores: A potential therapy for treatment of lipofuscin-based retinal diseases. Invest. Ophthalmol. Vis. Sci. 2005;46:4393-401.
21. Vogel S, Piantedosi R, O'Byrne SM, et al. Retinol-binding protein-deficient mice: Biochemical basis for impaired vision. Biochemistry 2002;41:15360-8.
22. Golczak M, Kuksa V, Maeda T, et al. Positively charged retinoids are potent and selective inhibitors of the trans-cis isomerization in the retinoid (visual) cycle. PNAS 2005;102:8162-7.
23. Fan J, Woodruff ML, Cilluffo MC, et al. Opsin activation of transduction in the rods of dark-reared Rpe65 knockout mice. J Physiol (Lond) 2005;568:83-95.
24. Batten ML, Imanishi Y, Tu DC, et al. Pharmacological and rAAV gene therapy rescue of visual functions in a blind mouse model of Leber congenital amaurosis. PLoS Med 2005;2:e333.
25. Donoso LA, Kim D, Frost A, et al. The role of inflammation in the pathogenesis of age-related macular degeneration. Surv Ophthalmol 2006;51:137-52.
26. Crabb JW, Miyagi M, Gu X, et al. From the Cover: Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. PNAS 2002;99:14682-7.
27. Johnson LV, Leitner WP, Staples MK, Anderson DH. Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp Eye Res 2001;73:887-96.
28. Mullins RF, Aptsiauri N, Hageman GS. Structure and composition of drusen associated with glomerulonephritis: Implications for the role of complement activation in drusen biogenesis. Eye 2001;15(Pt 3):390-5.
29. Thurman JM, Holers VM. The central role of the alternative complement pathway in human disease. J Immunol 2006;176:1305-10.
30. Edwards AO, Ritter R, III, Abel KJ, et al. Complement Factor H Polymorphism and Age-Related Macular Degeneration. Science 2005;308:421-4.
31. Haines JL, Hauser MA, Schmidt S, et al. Complement Factor H Variant Increases the Risk of Age-Related Macular Degeneration. Science 2005;308:419-21.
32. Klein RJ, Zeiss C, Chew EY, et al. Complement Factor H Polymorphism in Age-Related Macular Degeneration. Science 2005;308:385-9.
33. Hageman GS, Anderson DH, Johnson LV, et al. From The Cover: A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. PNAS 2005;102(:7227-32.
34. Magnusson KP, Duan S, Sigurdsson H, et al. CFH Y402H confers similar risk of soft drusen and both forms of advanced AMD. PLoS Med 2006;3:e5.
35. Gold B, Merriam JE, Zernant J, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet 2006;38:458.
36. Ambati J AA, Fernandez S, Sakurai E, Lynn BC, Kuziel WA, Rollins BJ, Ambati BK. An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nature Medicine 2003;9:1390-7.