The complement system consists of a series of soluble proteins derived from the liver that play a key role in eradication of foreign invaders. Activation of the system results in the production of the cytolytic membrane attack complex (MAC), leading to death of targeted cells via cell lysis. This system plays a key role in the prevention of infection by being involved in the recognition, opsonization and lysis of microorganisms and foreign pathogens. Complement activation can occur by one of three pathways: classical, lectin and alternate pathways.
The classical pathway is initiated when C1q (a pattern recognition molecule) binds to immunoglobulin in immune complexes, to C-reactive protein on self or microbial surfaces, or directly to molecules expressed on microbial membranes. This complex activates C1r, which becomes enzymatically active and cleaves C1s. C1s then activates C2 and C4, releasing C2a, C2b, C4a, and C4b. C2b and C4b become joined to form C3 convertase, C4b2b. C2 is similar (and related) to complement factor B (CFB), while C4 is similar (and related) to C3. The C4b2b molecule is functionally similar to C3bBb. C5 is the fifth component of complement, which plays an important role in inflammatory and cell killing processes. This protein is composed of alpha and beta polypeptide chains that are linked by a disulfide bridge. An activation peptide, C5a, which is an anaphylatoxin that possesses potent spasmogenic and chemotactic activity, is derived from the alpha polypeptide via cleavage with a convertase. The C5b macromolecular cleavage product can form a complex with the C6 complement component, and this complex is the basis for formation of the membrane attack complex, which includes additional complement components. The C5 convertase cleaves C5 into its two active components C5a and C5b. The result of this cleavage is the release of a C5a fragment, a potent inflammatory molecule, and activation of C5b, which initiates the MAC.
The MAC is initiated when the complement protein C5 convertase cleaves C5 into C5a and C5b. Another complement protein, C6, binds to C5b. The C5bC6 complex is bound by C7 and finally C8C8 alpha-gamma induces the polymerization of 10 to 16 molecules of C9 into the complex called MAC.
Initiation of the lectin pathway occurs when pattern recognition molecules (any of mannose-binding lectin (MBL), L-ficolin, H-ficolin, or M-ficolin) bind to the exterior surfaces of bacteria.
The alternative pathway is continuously activated by spontaneous hydrolysis of the internal thioester bond in C3 to form C3(H20). This molecule, although not cleaved, can fulfill the same role as C3b in C3 convertase. C3(H20) binds to CFB, and this complex is cleaved by the complement serine protease factor D (CFD), resulting in splitting of the CFB protein into Ba and Bb fragments. C3(H20)Bb is a C3 convertase, which splits C3 into C3a and C3b. Once produced by these means, C3b perpetuates the positive feedback loop described above, joining CFB to form more convertase enzyme. Newly cleaved C3b is also deposited on nearby surfaces. C3(H20)Bb is a less efficient enzyme than C3bBb, but is less easily inactivated by complement factor H (CFH) and its cofactor complement factor I (CFI).
Tight regulation of the complement cascade is necessary to prevent immune mediated disease and off-target damage of non-infected host cells. The balance between complement activation and inhibition is mediated by a series of regulatory proteins. CFH is critical inhibitor of the complement pathway that ensures the system targets foreign rather than host cells by neutralizing activated complement proteins that adhere to normal host cells.
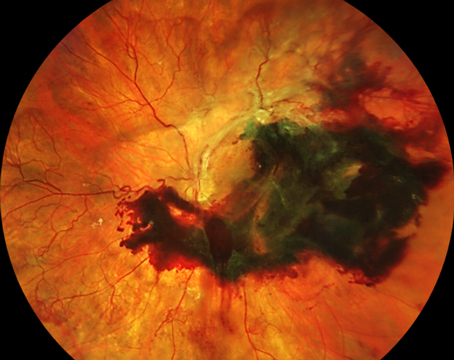
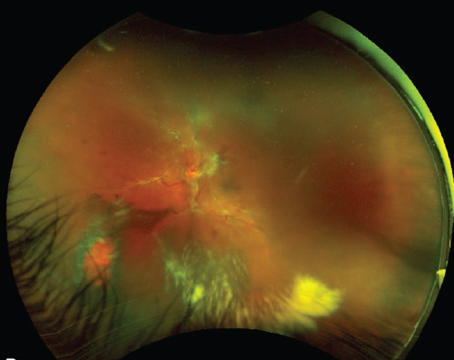
AMD and Complement
The leading cause of vision loss in industrialized countries is age-related macular degeneration and its prevalence is increasing with the aging of the population.1,2 The defect in AMD is thought to lie in the outer retina and retinal pigment epithelium, and genetic predisposition, age, ischemia and environmental factors may play a role in its development.3,4
Several recent studies have reported an association between AMD and key proteins in the complement cascade. For example, a variation in the gene encoding CFH, the key inhibitor of complement mediated damage to non-pathologic cells, produces a nearly fivefold increase in the risk of AMD for individuals who harbor the Y402H polymorphism.5-8 The products of complement activation have been detected in blood in AMD patients. Hendrik P. N. Scholl and colleagues found that C3d, CFB and CFD were significantly increased in patients with AMD.9 The presence of activation products in circulating blood indicates that the inflammation found in AMD is not limited to the retina, but is systemic. The extent of this systemic complement activation in AMD is currently unknown.
Mutations in complement-related genes including CFB, C2 and C3 have been associated with AMD.10 However, mutations in the CFH gene appear to have the greatest link to AMD.5-8 The CFH gene forms part of the RCA (regulators of complement activation) gene cluster located in the 1q32 chromosomal region of the human genome. The Y402H genetic variant of CFH gene is commonly associated with AMD. Genetic studies suggest that there may be further variants in the region associated with altered risk that have not yet been uncovered.
Local complement system in the human RPE-choroid complex has also been described. Cells within the neural retina, RPE and choroid demonstrated gene expression profiles consistent with replication of the complete cohort of complement components and regulatory proteins associated with both the classical and alternative pathways.11 Components of the complement system have also been identified in drusen.12,13 Druse in AMD contain almost all alternative complement pathway proteins, including CFH, C3, and the products of its activation and degradation, as well as the terminal pathway proteins C5, C6, C7, C8, C9, separately and in combination as the MAC. An implication of this hypothesis relates to the finding that homozygous individuals for the most common CFH risk allele (Y402H) tend to concurrently manifest raised levels of choroidal C-reactive protein, a serum biomarker for inflammation,14 and in patients with both problems an increased risk of AMD progression.15 Thus, since dysfunction of the local complement system is associated with AMD, the RPE-choroid complex is a potential location for the delivery of novel therapeutic agents that target the complement cascade.
Current Role of Complement-Based Therapies
A number of AMD therapies are being developed to target specific components of the complement cascade. Complement therapies also have the potential to work at an earlier stage in the disease process, preventing progression to late AMD. However, modulation of the complement cascade may adversely affect the body’s defense mechanisms. Hence, any new therapeutic modality must affect AMD without adversely affecting the body’s immunological function. The use of local complement component inhibitors minimizes potential systemic side effects of complement inhibition. However, local inhibition of complement via intravitreal injections may have a shorter half-life and more local adverse effects, and may not be feasible in a life-long disease such as AMD.
Some companies are evaluating nucleic acid aptamers that are synthetically derived and demonstrate desirable therapeutic properties largely owing to their three-dimensional structure, high target specificity, and high binding affinity. Others are using monoclonal antibodies that are laboratory-engineered clones of immunoglobulins designed to bind to specific target cell surfaces. They inhibit specific molecules or activate the immune system. Monoclonal antibody-based complement inhibitors exhibit similar molecular recognition characteristics, but they also have the potential to provoke an immunogenic response, which does not occur with aptamer compounds.
|
Some of the medications that are on the horizon and show potential in the management of AMD include:
• Eculizumab (Soliris, Alexion Pharmaceuticals, Cheshire, Conn.) is an orphan drug that is marketed for intravenous treatment of paroxysmal nocturnal hemoglobinuria (PNH), and received FDA approval in 2007 as the only licensed monoclonal antibody that targets the complement system. The molecule has been humanized to minimize immunogenicity and increase drug half-life, but was originally derived from a murine C5 antibody. This drug binds to C5 and prevents downstream activation and formation of MAC. The drug is administered intravenously over six months; weekly dosing during the initial induction period followed by two weekly maintenance doses. The COMPLETE (Complement inhibition of systemic Eculizumab for the treatment of Non-Exudative Age-Related Macular Degeneration) study at Bascom Palmer (Yehoshua Z, et al IOVS 2012;53:ARVO E-Abstract 2046) is one of the first to study the use of systemic complement inhibition for the treatment of dry AMD. (See Retinal Insider, September 2012 for more on the COMPLETE Trial.) Their results involving 30 eyes of 30 patients at six months showed that systemic treatment with eclizumab did not decrease drusen volume. In the same study, systemic use of eclizumab did not decrease the growth rate of geographic atrophy. Their explanations for lack of treatment effect for GA in this population included the possibility that complement activation may have no role in growth of GA; or the study duration was too short; or a higher drug dose was needed; inadequate penetration into the eye; different complement target needed; or that they used an inappropriate endpoint. These results were from a single study and involved administering systemic medication in just 30 eyes of 30 patients, and the power of the study may not be adequate to appropriately assess the role of C5 inhibition in dry AMD. Further studies are indicated in assessing the role of eclizumab in dry AMD.
• POT-4 (Alcon), a derivative of compstatin, is a potent C3 inhibitor that suppresses complement activation by preventing the formation of key elements within the proteolytic cascade, thus impeding local inflammation, upregulation of angiogenic factors and subsequent tissue damage. POT-4 is administered intravitreally, which may limit possible unwanted systemic effects. Early results of POT-4 suggest that it manifests a good safety profile and drug tolerability. (Kaushal S, et al IOVS 2009;50:ARVO e-abstract 5010.) It is currently in Phase II clinical trials.
• ARC1905 (Ophthotech, New York, N.Y.) is an aptamer-based C5 inhibitor, blocking the cleavage of C5 into C5a and C5b fragments and is another intravitreally administered complement inhibitor being evaluated in AMD. Like POT-4, it is similarly selective for a centrally positioned component within the cascade, although exerting its effect further downstream.
• FCFD4514S (Genentech) is an anti-factor D antibody being studied for the treatment of dry AMD. Blockage of the complement factors (such as factor D) that moderate the production of these end-products serves to attenuate complement activation, rather than shutting the system down completely. Anti-factor D is administered intravitreously and selectively inhibits CFD which is a key component of the amplification step of the alternatve pathway. It is currently in Phase II testing for dry AMD.
• TA106 (Taligen Therapeutics; Alexion Pharmaceuticals, Cheshire, Conn.) is a CFB inhibitor, still in pre-clinical development, that is primarily being investigated as an inhaled formulation in the treatment of severe, chronic asthma refractory to current therapies and is recently being studied for macular degeneration.16
• JSM-7717 (EvaluatePharma) and JPE-1375 (Jerini AG; Berlin, Germany) are two peptidomimetic C5a receptor (which is pro-inflammatory) antagonists currently in preclinical assessment for AMD.17 Receptor antagonists competitively bind to the C5a receptor neutralizing interaction, and hence such drugs have the potential to suppress the inflammatory response without adversely affecting complement-related immunity and are being used in pre-clinical studies.18
• Intravenous administration of CR2-fH, a recombinant form of CFH, was associated with a reduction in CNV size and the physiological sequelea of CNV on retinal function. The effectiveness of this approach in a mouse model of laser-induced AMD was recently elucidated.19 This strategy involves the supplementation of wild-type CFH with a recombinant “protective” form of the protein in high-risk variant cases. In theory, such augmentation should be helpful in re-instituting homeostatic regulation of the alternative pathway of the complement system. This drug is being studied in animal models and is part of ongoing pre-clinical experiments.
• C1INH (ViroPharma Inc., Exton, Pa.) received FDA approval for the treatment of hereditary angioneurotic edema in 2008. Mutations in the gene that encodes SERPING1, a C1-inhibitor (C1INH) may similarly play a role in the development of AMD.20 A recent study showed that C1INH is present in the human retina and choroid, and AMD affection status was correlated with increased levels of the protein in the choroid.20 This drug, although not being used clinically, has the potential to be used in AMD.
Thus, there is substantial evidence now that the complement system plays a role in the pathogenesis of AMD, but its exact role is uncertain. A number of drugs are being developed that target various components of the complement cascade, predominantly the alternate complement pathway. The complement cascade remains as one of the many therapeutic targets in AMD, and our understanding of its role in AMD is still primitive. However, emerging drugs that target the complement cascade are promising and may play a vital role in the prevention and management of this condition in the future.
Dr. Kaiser is a consultant to Alcon, Novartis, Genentech, Ophthotech, Allegra, Bayer, Regeneron and ArcticDx; he reports a financial interest in SKS Ocular, LLC. Contact him at Cole Eye Institute, 9500 Euclid Ave., Desk i3. Cleveland, Ohio 44195. Phone: (216) 444-6702, e-mail:
pkkaiser@aol.com.
1. La Cour M, Kiilgaard JF, Nissen MH. Age-related macular degeneration: Epidemiology and optimal treatment. Drugs Aging 2002;19:101-133.
2. Klein R, Klein BE, Lee KE, Cruickshanks KJ, Gangnon RE. Changes in visual acuity in a population over a 15-year period: The Beaver Dam Eye Study. Am J Ophthalmol 2006;142:539-549.
3. Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science 2005;308:385-389.
4. Mori K, Horie-Inoue K, Kohda M, et al. Association of the HTRA1 gene variant with age-related macular degeneration in the Japanese population. J Hum Genet 2007;52:636-641.
5. Hageman GS, Anderson DH, Johnson LV et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA 2005;102:7227-32.
6. Klein RJ, Zeiss C, Chew EY et al. Complement factor H polymorphism in age-related macular degeneration. Science 2005;308:385-9.
7. Edwards AO, Ritter RR, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science 2005;308:421-4.
8. Haines JL, Hauser MA, Schmidt S et al. Complement factor H variant increases the risk of age-related macular degeneration. Science 2005;308:419-21.
9. Scholl HP, Charbel Issa P, Walier M, Janzer S, Pollok-Kopp B, Borncke F et al. Systemic complement activation in age-related macular degeneration. PLoS One 2008;3:e2593.
10. Yates JR, Sepp T, Matharu BK et al. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med 2007;357:553-61.
11. Anderson DH, Radeke MJ, Gallo NB et al. The pivotal role of the complement system in aging and age-related macular degeneration: Hypothesis re-visited. Prog Retin Eye Res 2010;29:95-112.
12. Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res 2001;20:705-732.
13. Persson BD, Schmitz NB, Santiago C, Zocher G, Larvie M, Scheu U et al. Structure of the extracellular portion of CD46 provides insights into its interactions with complement proteins and pathogens. PLoS Pathog 2010; 6:e1001122.
14. Johnson PT, Betts KE, Radeke MJ, Hageman GS, Anderson DH, Johnson LV. Individuals homozygous for the age-related macular degeneration risk-conferring variant of complement factor H have elevated levels of CRP in the choroid. Proc Natl Acad Sci USA 2006;103:17456-61.
15. Robman L, Baird PN, Dimitrov PN, Richardson AJ, Guymer RH. C-reactive protein levels and complement factor H polymorphism interaction in age-related macular degeneration and its progression. Ophthalmology 2010;117:1982-88.
16. Taube C, Thurman JM, Takeda K et al. Factor B of the alternative complement pathway regulates development of airway hyperresponsiveness and inflammation. Proc Natl Acad Sci USA 2006;103:8084-8089.
17. Schnatbaum K, Locardi E, Scharn D et al. Peptidomimetic C5a receptor antagonists with hydrophobic substitutions at the C-terminus: Increased receptor specificity and in vivo activity. Bioorg Med Chem Lett 2006;16:5088;92.
18. Allegretti M, Moriconi A, Beccari AR et al. Targeting C5a: Recent advances in drug discovery. Curr Med Chem 2005;12:217-36.
19. Rohrer B, Long Q, Coughlin B et al. A targeted inhibitor of the alternative complement pathway reduces angiogenesis in a mouse model of age-related macular degeneration. Invest Ophthalmol Vis Sci 2009;50:3056-64.
20. Mullins RF, Faidley EA, Daggett HT, Jomary C, Lotery AJ, Stone EM. Localization of complement 1 inhibitor (C1INH/SERPING1) in human eyes with age-related macular degeneration. Exp Eye Res 2009;89:767-73.s.