The craniosynostoses are autosomal dominant birth defects caused by premature fusion of one or several cranial sutures in utero, resulting in restricted skull growth and brain development as well as a variety of other phenotypic dysmorphisms. These anomalies can range from maxillary hypoplasia to syndactyly with psittichorhina. The ocular consequences of craniosynostoses include malformed, shallow orbits, leading to exophthalmos/proptosis. These malformations can lead to vision-threatening sequelae from possible amblyopia, strabismus, exposure keratopathy and potential optic nerve injury. In this article, we’ll describe the causes, types and manifestations of craniosynostosis, as well as monitoring and treatment recommendations.
Causes and Genetics
Craniosynostosis is associated with more than 180 different syndromes. Crouzon Syndrome (CS), Apert Syndrome (AS) and Pfeiffer Syndrome (PS) are the most prevalent. While their severities, phenotypes and ocular manifestations differ slightly, all three disorders stem from mutations in the Fibroblast Growth Factor Receptor genes. Each of the four FGFR genes is responsible for producing a fibroblast growth factor receptor protein, a membrane-spanning tyrosine kinase receptor whose signaling cascade directs important functions such as cell proliferation, angiogenesis and, notably, osteogenesis.
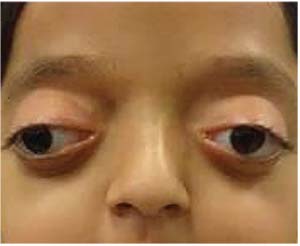 |
| Figure 1. Patient with Crouzon Syndrome with typical facies. Note exotropia, excyclorotation of the orbits and mild ptosis. |
The mutations associated with these three syndromes are, predictably, gain-of-function mutations in the FGFR genes. With these changes, FGFR proteins become better ligand binders—hyperactive, or ligand-independent, receptors that result in an abnormally quick ossification of the cranial sutures. Although genetic analysis has made great strides in identifying the precise mutations responsible for each syndrome, especially with AS, in which 98 percent of cases stem from one of two point mutations,1 the vast majority of FGFR-related craniosynostosis syndromes are examples of allelic heterogeneity. Thus, clinically the focus is often on phenotype instead of specific genotype.
Clinical Presentations
The mutations are completely penetrant, meaning all carriers have traits, yet the carriers exhibit variable expressivity. Therefore, clinical presentations can vary. Crouzon Syndrome is the most common craniosynostosis and is the mildest FGFR-related craniosynostosis phenotypically. The degree of facial deformity is often less severe and there is no involvement of the extremities. First described in 1912, the Crouzon disease triad consists of calvarial deformities, facial anomalies and proptosis,2 and patients rarely exhibit developmental delay.
Conversely, patients with Apert and Pfeiffer Syndromes can have mild to moderate intellectual disabilities and extremity involvement, in addition to the facial deformities exhibited in CS. AS patients often present with syndactyly in hands and feet, and PS patients possess variable brachydactyly and broad, medially deviated thumbs and toes.
The wide range of clinical presentations of these syndromes is due to the variable nature of the cranial sutures involved in each case. Although early fusion of the coronal suture is the most common manifestation, CS, AS and PS often exhibit complex or even pansynostosis, involving several if not all sutures. Such fusion means growth can’t occur in a direction perpendicular to the suture, thus limiting the skull’s development anteriorly and leading to the common anatomical characteristics shared by the three syndromes: hypertelorism; proptosis; and midface hypoplasia. (Figures 1 and 2)
Ocular Manifestations
The complications due to facial hypoplasia and shallow orbits are numerous and consistent between the different syndromes; however, severity will vary. Although there is some evidence that FGFR2 may have a direct role in ocular anterior chamber development,3 the ophthalmic sequelae of craniosynostosis is truly due to deficient anterior calvarial growth, midfacial hypoplasia and increased intracranial pressure.
The most common cause of decreased visual acuity in these patients is proptosis-related strabismus leading to amblyopia.4 Shallow orbits increase the risk of exocyclorotated orbits and, therefore, aberrant insertion or even malformation of the extraocular muscles. This orbital abnormality often results in incomitant strabismus. Exotropia is the most common horizontal manifestation. The combination of shallow orbits, lack of inferior support due to midface hypoplasia and orbital floor recession results in overaction of the inferior oblique, leading to vertical misalignments. Collectively, this can manifest in V-pattern strabismus and, if untreated, eventual amblyopia.
However, amblyopia can be strabismus-independent and instead due to undiagnosed refractive errors and anisometropia. These patients exhibit higher rates of ametropia and astigmatism, contributing to a higher occurrence of refractive amblyopia.
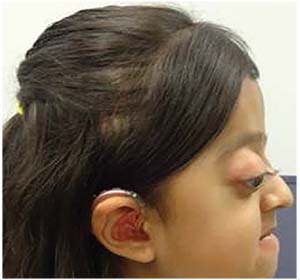 |
| Figure 2. Profile view highlighting shallow orbits with exophthalmos and proptosis, psittichorhina, and maxillary hypoplasia. |
Another cause of amblyopia can be related to lid ptosis observed alongside superior rectus hypoplasia and malformation of the superior levator palpebrae muscle. These patients may adopt a chin-up posture in an effort to compensate for ptosis. These abnormalities can cause deprivation amblyopia.
In addition, vision may be affected by optic nerve injury that can result from either severe exophthalmos causing tension, increased intracranial pressure or a combination of the two. Shallow orbits (Figure 2) can also put the patient at risk for globe subluxation or herniation of the globe itself, contributing to optic nerve injury.
Lastly, corneal scarring from exposure keratopathy can be a major concern. Regular monitoring of the ocular surface for breakdown and dryness is required. This can occur with even small degrees of craniosynostosis-related proptosis. Whether it’s from structural exposure, lid abnormalities or nasolacrimal dysfunction, the corneal epithelium can be at risk. Resulting scarring or surface irregularities can lead to refractive or deprivation amblyopia with permanent vision loss.
Diagnosis
Craniosynostosis syndromes like CS, AS and PS may be detectable prenatally by noting an aberrant skull shape and can be confirmed with a family history and genetic testing for FGFR mutations. Children may not present with proptosis immediately at birth, but the symptoms are progressive and will likely develop within the first years of life. Thus, the role of the ophthalmologist, initially, is to perform early, complete exams. Amblyopia should be monitored and it’s recommended that the patient undergo refraction with cycloplegic retinoscopy. Regular dilated fundus exams are recommended to monitor for possible optic atrophy. If the patient is cooperative and able, visual field testing should be used to detect any loss of peripheral vision, and optical coherence tomography can visualize and monitor the optic nerves.
Treatment
There is no singular treatment to remedy all of the above symptoms, yet surgical release of the involved sutures in the first year of life can significantly improve brain development and reduce midface hypoplasia and proptosis. Increasing cranial volume and normalizing intracranial pressure are often the foremost goals of early treatment. Surgical treatment drastically reduces the risk of optic atrophy due to increased intracranial pressure.
Regardless of concomitant manifestations of the syndrome, amblyopia, whether secondary to refractive error, deprivation, strabismus or a combination, should be treated as early as possible after confirmation via a complete eye exam. For refractive causes, proper correction should be given in the form of spectacles. If a difference in vision persists even with proper refractive correction, patching or atropinization of the dominant eye may be necessary. It’s critical to start early and work to maintain vision until major craniofacial surgeries are completed and any necessary strabismus surgeries are performed.
Ideally, strabismus surgery should be performed at the earliest possible time to facilitate the development of proper vision and binocularity; however, if surgery to advance the midface or adjust the orbit is anticipated, strabismus surgery should be delayed six months to a year,5,6 as such craniofacial surgery will alter the orbital volume and ability to achieve perfect alignment.
When strabismus surgery is performed, it can be complicated due to the wide range of anatomical anomalies. Surgery should be tailored to the specific misalignment with a goal of orthophoria and improved binocularity. Again, particular attention must be paid to alignment in side gaze due to vertical muscle dysfunction.
Additionally, steps should be taken early to address any signs of exposure keratopathy. Treatment often consists of lubrication and taping shut the eyes at night. Associated ptosis can be protective and prevent corneal breakdown despite significant proptosis. If the proptosis does result in exposure keratopathy, temporary partial lateral tarsorrhaphy or orbital decompression may be necessary. The latter also has the added benefit of further alleviating the possible risk of compressive optic nerve injury.
In conclusion, the ophthalmic concerns for a patient with craniosynostosis are extensive and variable, and early, full exams should be performed to catch and assess amblyopia and other symptoms as soon as possible. However, whether a child presents with Crouzon, Apert, Pfeiffer or another such syndrome, complete management demands a multidisciplinary approach. The subspecialists involved should include ophthalmologists, craniofacial surgeons, geneticists, pediatric maxillofacial surgeons and, possibly, psychologists and therapists. REVIEW
Ms. Salevitz is a medical student at the University of Arizona College of Medicine.
Dr. Fecarotta is an attending physician and Dr. Singh is chief of the plastic surgery division at Phoenix Children’s Hospital.
1. Ferreira JC, Carter SM, Bernstein PS, et al. Second-trimester molecular prenatal diagnosis of sporadic Apert syndrome following suspicious ultrasound findings. Ultrasound Obstet Gynecol 1999;14:426–30.
2. Crouzon F. Dysostose cranio-faciale hereditaire. Bull Mem Soc Med Hop Paris 1912;33:545–555.
3. K. Okajima, L.K. Robinson, M.A. Hart, D.N. Abuelo, L.S. Cowan, T. Hasegawa, et al. Ocular anterior chamber dysgenesis in craniosynostosis syndromes with a fibroblast growth factor receptor 2 mutation, Am J Med Genet 1999;85:160-170.
4. Gray TL, Casey T, Selva D, et al. Ophthalmic sequelae of Crouzon syndrome. Ophthalmology 2005;112:1129–1134.
5. G.R. Diamond, L. Whitaker. Ocular motility in craniofacial reconstruction Plast Reconstr Surg 1984;73:31-37.
6. Morax S. Change in eye position after cranio-facial surgery. J Maxillofac Surg 1984;12:47-55.


