|

This first installment of a two-part series will review the conditions that mimic dry AMD. Next month, the focus will shift to wet AMD.
Local inflammation and activation of the complement cascade have been implicated in drusen formation. The central role of polygenic risk factors, cellular oxidative stress, complement system pathways and local inflammatory processes in AMD pathogenesis are widely supported by experimental, clinical and epidemiological evidence.3-9
|

The widely variable clinical presentation and sometimes unpredictable natural history are important confounding features in the correct diagnosis of AMD.10 Variable degenerative and dystrophic diseases of the retina and RPE, in which pigmentary mobilization or lipofuscin accumulation are major findings, share fundamental clinical features with the dry form of the disease. Novel diagnostic tools such as fundus autofluorescence imaging have led to an increasing understanding of pathophysiologic mechanisms in retinal degeneration. FAF is a non-invasive imaging technique that enables the visualization of RPE and photoreceptor cell layer changes, registering the retinal intrinsic fluorescence derived from lipofuscin.11 Epidemiological data and complimentary systemic or genetic tests are sometimes required. As in dry AMD, therapeutic benefits are currently limited in most of its mimicking conditions. However, the specific diagnosis should be determined due to the high variability in visual prognosis among those different entities.
Best's Disease
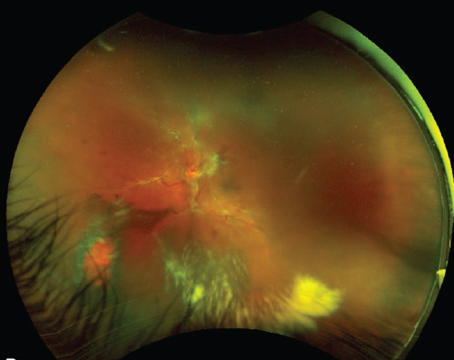
Best's disease, or vitelliform macular dystrophy type 2, is a rare autosomal dominant disease with variable penetrance and expressivity. Careful review of the family history may be helpful in making the diagnosis. The typical egg-yolk lesion in the central macula tends to evolve from this typical "vitelliform stage" to the "pseudo-hypopyon stage," then to the "vitelliruptive stage" (See Figure 1). Visual impairment in general progresses after the juvenile onset of the disease. Functional and morphological stabilization usually occur one or two decades after initial presentation, when eventually all of the yellow pigment may disappear leaving a circumscribed area of atrophic RPE. The underlying pathophysiology is still poorly understood.11,12
The clinical presentation of Best's disease may vary widely, and sometimes the differential diagnosis is difficult. Mild forms may present lipofuscin deposition and pigmentary mobilization in the fovea as major clinical features, resembling dry AMD. Severe or complicated forms of Best's disease, in the late stages, are particularly difficult to differentiate from wet AMD solely on a clinical basis. Abnormal electrooculographic (EOG) findings are universally present in patients with Best's disease regardless of the clinical presentation, and are thus decisive in the differential diagnosis.
|


Pattern Dystrophies of the RPE
Autosomal dominant pattern dystrophies of the RPE are characterized by variable patterns of pigment deposition in the macular area, associated with the development of mild disturbances of central vision. These entities typically occur in midlife and carry a good visual prognosis in at least one eye. Although the electroretinogram is generally normal, the EOG can be slightly or moderately subnormal.12 Pattern dystrophies of the RPE are usually inherited as an autosomal dominant trait, and mutations of the peripherin/retinal degeneration slow (RDS) gene have been demonstrated in some families.
Pattern dystrophies have been subdivided into a few principal groups based on the distribution of pigmentary changes in the fundus. However, this distinction can be difficult to establish in clinical practice when fundus findings do not precisely agree with any classification. The observed patterns may be different in the two eyes of one patient; a patient may show progression from one pattern to another over the years, and some pedigrees show unpredictable combinations of the main patterns.12
According to Donald Gass's classification, the principal sub-groups of pattern dystrophies are:
- Group 1: adult-onset vitelliform macular dystrophy (AVMD, See Figure 2);
- Group 2: butterfly-shaped pigment dystrophy;
- Group 3: reticular dystrophy of the RPE;
- Group 4: multifocal pattern dystrophy, (See Figure 3); and
- Group 5: coarse pigment mottling in the macula (fundus pulverulentus).
The typically associated lipofuscin deposition may resemble fundus flavimaculatus, especially if widespread in the macular area. Clinical presentations that evolve with RPE atrophy are particularly difficult to differentiate from dry AMD. Choroidal neovascularization is rare, although it may develop in any subtype of these entities.13 When it occurs, the differential diagnosis between the macular lesion and wet AMD may be challenging.
Cuticular Drusen, Pseudo-Vitelliform Detachment
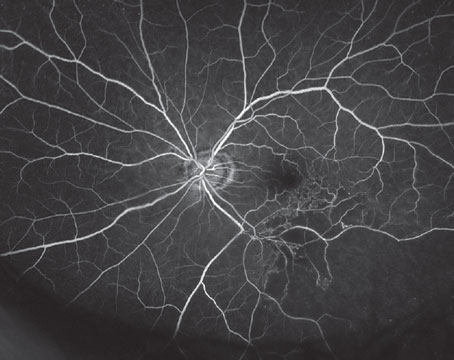
Cuticular drusen, or basal laminar drusen, with the associated pseudo-vitelliform macular detachment, was first described by Donald Gass. In the classic presentation of the so-called "Gass syndrome," numerous small, uniformly sized, round, yellow, sub-retinal nodules are arranged in clusters in the posterior pole. Cuticular drusen appear in the early phase of fluorescein angiography in a characteristic pattern, described as "stars in the sky."12,14 Concomitant occurrence of soft drusen is frequent, which may coalesce into the characteristic yellow macular deposition and associated serous neurosensory retinal detachment, resembling the vitelliform stage of Best's disease (See Figure 5).14 The condition usually leads to preserved visual acuity until late in life, and normal electrophysiologic tests are the rule. It may rarely evolve to geographic atrophy and choroidal neovascularization.12
Fundamental clinical similarities between Gass's syndrome, adult onset vitelliform macular dystrophy and other pattern dystrophies of the RPE have raised the question of whether they are all variants of the same disease. However, genetic screening for the known disease-causing genes in all these entities has shown no association to the syndrome. This phenotype also shares with AMD several epidemiological, clinical, and histopathological features.
Stargardt's Disease
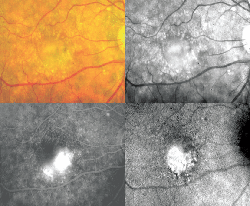
Stargardt's disease is the most common hereditary macular dystrophy and accounts for approximately 7 percent of retinal degenerative diseases. It is characterized by bilateral atrophy of the macula and underlying RPE, associated central vision impairment (See Figure 6), and frequent presence of prominent flecks in the posterior pole of the retina known as fundus flavimaculatus (See Figure 7).
Many authors believe that Stargardt's disease and fundus flavimaculatus are actually different spectrums of the same disease.12,14
|
Stargardt's disease is most commonly inherited as an autosomal recessive trait, and a recessive locus has been identified on chromosome 1p13-p22 (STGD1), mostly related to mutations in the ABCA4 gene (previously called ABCR gene). Fundus flavimaculatus associated genetic changes were identified in the same chromosome region allelic to STGD1. Autosomal dominant loci of Stargardt's have been mapped to chromosomes 13q (STGD2), 6q11-14 (STGD3), and 4p (STGD4).
The typical finding on fluorescein angiography described as the "dark choroid" can contribute to the diagnosis of Stargardt's disease, although it may not be observed in the majority of the cases. Fundus autofluorescence is not universally high in STGD/FFM. Different presentations have been described, as well as the correlation of autofluorescence levels and functional or electroretinographic abnormalities.16 Interestingly, in ABCA4-associated retinal diseases, a peripapillary ring-shaped region of normal-appearing autofluorescence has been described in all stages of Stargardt's disease, which may aid in the recognition of these entities.17
Central Serous Chorioretinopathy
Central Serous Chorioretinopathy manifests in the acute stage as a serous elevation of the neurosensory retina occasionally associated with small retinal pigment epithelial detachments in the macula of unknown cause. While extensive demographic data indicate that CSC is predominantly an ailment of young to middle-aged men, commonly triggered by emotional stress,18 CSC can also occur in women, in the elderly and, rarely, in children.19
Symptoms are generally nonspecific and transitory; and the condition may even be asymptomatic with the diagnosis being made in routine examinations at a later time, based on retinal pigment epithelium changes such as mobilization. Bilateral involvement is estimated in 4 percent to 20 percent of the cases, and may be more frequent when underlying endogenous or exogenous hypercortisolism is present.20
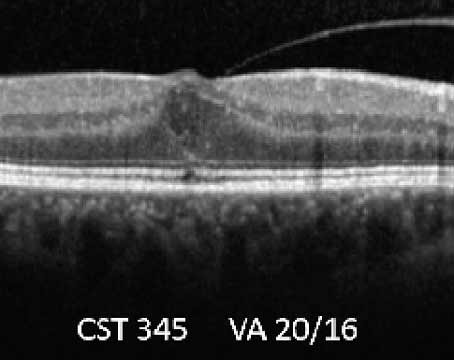
The diagnosis of CSC is usually based on clinical findings supported by demographic characteristics and medical history. Additional testing is helpful in the differential diagnosis of atypical cases or if secondary complications are suspected. Whereas the pigmentary changes associated with CSC may mimic dry AMD if they affect the central macula (See Figure 8), wet AMD must be ruled out due to the exudative nature of the condition. Focal leaks at the level of the RPE may be evident on fluorescein angiography, and regions of choroidal hyperpermeability are revealed in indocyanine green angiography.21 The classic "smokestack" pattern of fluorescein angiographic leakage is present only in approximately 10 percent of acute cases.22 The serous retinal detachment and associated RPE detachments are evident on optical coherence tomography of the macula. Small serous PEDs are frequently detected on OCT in eyes with asymptomatic or quiescent disease and also in contra-lateral eyes. Fundus autofluorescence imaging is of particular value in the diagnosis of CSC, as it clearly highlights the gravitational pattern of subretinal leakage characteristic of the disease.23,24
|
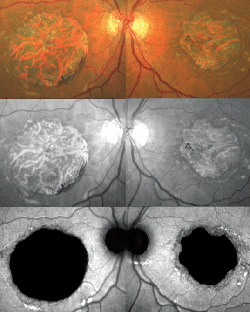
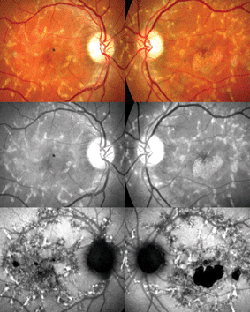
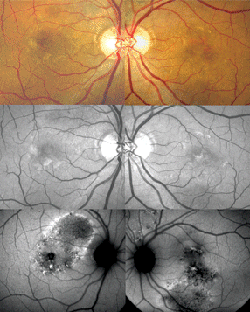
Clinical manifestations tend to resolve spontaneously in approximately three to six months, but recurrences are expected to occur in up to 50 percent of patients. The functional prognosis is typically good, although chronic or recurrent cases may evolve with visual impairment.23 A worse visual outcome may also be observed in association with complications such as bullous retinal detachment, subretinal fibrosis or secondary choroidal neovascularization.25 Secondary choroidal neovascularization is the most visually threatening complication in CSC. CNV may occur as part of the natural course of the disease or secondary to thermal laser photocoagulation and possibly PDT.
AMD is the leading cause of blindness in developed countries, but many other entities may have similar manifestations. Variable dystrophic or degenerative conditions of the retina share with dry AMD characteristic features involving pigmentary disturbances and lipofuscin deposition. Some demographic data and clinical findings may be of special value in the differential diagnosis. Dry AMD-mimicking conditions should be suspected especially in individuals younger than 50 years old and when pathological changes extend beyond the central macular area.
Nevertheless, the differential diagnosis is often challenging and sometimes impossible on a clinical basis. New fundus imaging techniques, such as fundus autofluorescence and spectral domain OCT, may be especially helpful in revealing fundamental diagnostic features of these dry AMD mimickers. The identification of disease-causing genes has further contributed to our understanding of underlying disease mechanisms of both AMD and these variable mimicking conditions. Identification of these other diseases is instrumental in determining the visual prognosis. The establishment of genotypic/phenotypic correlations may lead us to reliable diagnostic tools and potential therapeutic targets. Innovations such as genetic therapy or RPE cell transplantation are currently under development and are expected to be available for clinical tests in the near future.
Supported by The Macula Foundation, Inc. The authors have no financial interest in this report.
Dr. Ferrara did a retina fellowship at the LuEsther T. Mertz Retinal Research Center, New York City, and is a now doctoral candidate in the the Department of Ophthalmology, Otolaryngology and Head and Neck Surgery at the University of Sao Paolo. Drs. Freund and Yannuzzi practice at Vitreous-Retina-Macula Consultants of New York, and Dr. Yannuzi is in the Department of Ophthalmology at Columbia University. Contact Dr. Freund at 460 Park Ave. 5th floor, New York, NY 10022. Phone: (212) 861 979, fax: (212) 628 0698, e-mail:
mail@ vrmny.com.
1. Klein ML, Ferris III FL, Armstrong J, et al. AREDS Research Group. Retinal precursors and the development of geographic atrophy in age-related macular degeneration. Ophthalmology 2008;115:1026-1031.
2. Bird AC, Bressler NM, Bressler SB, et al. An international classification and grading system for age-related maculopathy and age-related macular degeneration. Surv Ophthalmol 1995;39:368-374.
3. Allikmets R, Shroyer NF, Singh N, et al. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science 1997;277:1805-1807.(a)
4. Klein R, Zeiss C, Chew EY, et al. Complement Factor H polymorphism in age-related macular degeneration. Science 2005;308:385-389.
5. Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science 2005;308:419-421.
6. Edwards AO, Ritter R 3rd, Abel KJ, et al. Complement factor H polymorphism and age-related macular degeneration. Science 2005;308:421-424.
7. Jakobsdottir J, Conley YP, Weeks DE, et al. Susceptibility genes for age-related maculopathy on chromosome 10q26. Am J Hum Genet 2005;77:389-407.
8. Gold B, Merriam JE, Zernant J, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet 2006;38:458-462.
9. Moshfeghi DM, Blumenkranz MS. Role of genetic factors and inflammation in age-related macular degeneration. Retina 2007;269-275.
10. Kalayoglu MV, Miller JW. Infection, inflammation and age-related macular degeneration. Clin Experiment Ophthalmol 2007;35:3-4.
11. Spaide RF, Noble K, Morgan A, Freund KB. Vitelliform macular dystrophy. Ophthalmology 2006;113: 1392-1400.
12. Gass JDM. Stereoscopic atlas of macular diseases: Diagnosis and treatment. 4th ed. St. Louis: Mosby, 1997:52-70.
13. Feist RM, White MF Jr, Skalka H, Stone EM. Choroidal neovascularization in a patient with adult foveomacular dystrophy and a mutation in the retinal degeneration slow gene (Pro 210 Arg). Am J Ophthalmol 1994;118:259-260.
14. Barbazetto IA, Yannuzzi NA, Klais CM, et al. Pseudo-vitelliform macular-detachment and cuticular drusen: Exclusion of 6 candidate genes. Ophthalmic Genet 2007;28: 192-197.
15. Allikmets R, Shroyer NF, Singh N, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet 1997;15:236-246.(b)
16. Lois N, Halfyard AS, Bird AC, Holder GE, Fitzke FW. Fundus autofluorescence in Stargardt macular dystrophy-fundus flavimaculatus. Am J Ophthalmol 2004;118: 55-63.
17. Cideciyan AV, Swider M, Alemn TS, et al. ABCA4-associated retinal degenerations spare structure and function of the human parapapillary retina. Invest Ophthalmol Vis Sci 2005;46(12):4739-4746.
18. Yannuzzi LA. Type A behavior and central serous chorioretinopathy. Retina 1987;7:111-131.
19. Spaide RF, Campeas L, Haas A, et al. Central serous chorioretinopathy in younger and older adults. Ophthalmology 1996;103:2070-2080.
20. Bouzas EA, Karadimas P, Pournaras CJ. Central serous chorioretinopathy and glucocorticoids. Surv Ophthalmol 2002;47:431-448.
21. Guyer DR, Yannuzzi LA, Slakter JS, Sorenson JA, Ho A, Orlock D. Digital indocyanine-green videoangiography of central serous chorioretinopathy. Arch Ophthalmol 1994;112:1057-1062.
22. Yamada K, Hayasaka S, Setogawa T. Fluorescein-angiographic patterns in patients with central serous chorioretinopathy at the initial visit. Ophthalmologica 1992;205:69-76.
23. Spaide RF, Klancnik JM. Fundus autofluorescence and central serous chorioretinopathy. Ophthalmology 2005;112:825-833.
24. Eandi CM, Ober M, Iranmanesh R, Peiretti E, Yannuzzi LA. Acute central serous chorioretinopathy and fundus autofluorescence. Retina 2005;25:989-993.
25. Loo RH, Scott IU, Flynn HW Jr, Gass JD, et al. Factors associated with reduced visual acuity during long-term follow-up of patients with idiopathic central serous chorioretinopathy. Retina 2002;22:19-24.