During development, this fetal vasculature nourishes the developing lens and vitreous.1 Any abnormalities along the involutional pathway produce the various manifestations of PFV. These are typically categorized as anterior, posterior or combined. Within the differential diagnosis of congenital leukocoria, PFV must be distinguished from retinoblastoma and retinopathy of prematurity, along with other potentially serious conditions. The posterior subtype has historically had very poor surgical outcomes and visual results.
PFV typically presents as a unilateral, idiopathic congenital malformation. Morton F. Goldberg, MD, in the 1997 Edward Jackson Memorial Lecture, introduced the term “persistent fetal vasculature” to replace “persistent hyperplastic primary vitreous,” which he felt was a misnomer because of its failure to include all of the fetal intraocular vasculature, rather than just the post-lental vessels. PFV indicates that at least partial, and possibly total, persistence of this intraocular vasculature remains after birth.2 Since the introduction of this broader and more inclusive definition, clinicians have been better able to stratify the disease entity and manage its various manifestations.
Embryogenesis
The hyaloid vascular system begins to form in the fourth to fifth week of gestation when the hyaloid artery enters the optic cup inferiorly from the primitive dorsal ophthalmic artery, a branch of the internal carotid. It proceeds anteriorly and extends to the posterior pole of the lens. The vasa hyaloidea propria branches from the hyaloid artery into the vitreous cavity; in correlation, the tunica vasculosa lentis, a capillary network also branching from the hyaloid artery, covers the lens surface by the time the fetus is 8 to 9 mm in length.3 The posterior aspect of this latter network connects to the choroidal vasculature through the annular vessel at the anterior border of the fetal optic cup, forming the iridohyaloid vessels. These vessels extend radially alongside the equator of the lens.2 The pupillary membrane is made up of anterior progression of the tunica vasculosa lentis. Along with fibrils and mesenchymal cells, this vasculature forms the primitive vitreous by two to three months gestation (40 to 60 mm in length). Of note, the hyaloid vasculature contains no veins. All drainage occurs through the choroidal vessels.
|
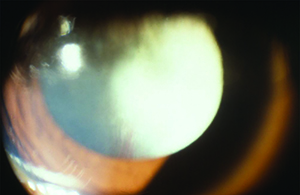
Clinical Findings
Persistence of any of the fetal vasculature elements can be an isolated phenomenon, or can be found in combination; however, dividing the pathology into anterior, posterior or combined subtypes affords the clinician and patient therapeutic and diagnostic dichotomies. Dr. Adrian Hunt and colleagues defined the anterior subtype as the presence of a retrolental opacity, elongated ciliary processes or cataract. Posterior PFV included findings of an elevated vitreous membrane from the optic nerve, retinal fold or dysplasia, retinal detachment or optic nerve hypoplasia.7
Persistent fetal vasculature can also be seen in conjunction with other ocular abnormalities, including morning glory disc anomaly, Peter’s anomaly, macular hypoplasia, microcornea and microphthalmos.2 PFV is usually unilateral, and one case series by Zane F. Pollard, MD, and colleagues described 84 percent of his PFV patients with unilateral microphthalmos and only 2 percent with bilateral disease. PFV associated with systemic syndromes often is bilateral, but in this case series, those patients with bilateral disease had no further systemic findings.5 Notable syndromes associated with PFV include: Norrie’s disease, which presents with retinal dysplasia, optic nerve hypoplasia, deafness and mental retardation; trisomy 13 (Patau syndrome) with clinical findings including cleft lip or palate, polydactyly and heart defects; and the Walker-Warburg syndrome, which can present with hydrocephalus, agyria, retinal dysplasia and congenital retinal nonattachment.2
Dr. Pollard also found that 3 percent of full-term children have some clinically detectable remnants of the hyaloid system.5 Most commonly, a persistent pupillary membrane can sometimes be observed on clinical exam as threadlike remnants of the anterior tunica vasculosa lentis. Occasionally described as pigmented “stars” on the anterior lens surface, this entity can also be seen causing pupil deformity and congenital ectropian or entropion uveae. If these vascular remnants are still perfused, spontaneous hyphema may occur. The most severe form of persistent pupillary membrane causes complete obscuration of the pupil with subsequent reduced vision and potential amblyopia.2
During development, regression of the iridohyaloid vessels allows for growth of the zonular ligaments. With persistence of the iridohyaloid vasculature, the clinician may see superficial vessels in the iris stroma with possible hairpin loops near the pupillary sphincter. If, subsequently, the zonules do not develop properly, lens subluxation may occur, typically away from the persistent vessel.2 Persistent iridohyaloid arteries have also been associated with absence of the fovea and consequently, poor vision.2 These abnormally persistent vessels can also be identified using fluorescein angiography. Elongated ciliary processes secondary to traction of the fibrovascular membrane are a common finding in anterior PFV, and were once thought to be pathognomonic.
A shallow anterior chamber can also be found and can even progress to fulminant angle-closure glaucoma. The etiology includes peripheral anterior synechiae, posterior synechiae and anterior displacement of the lens-iris diaphragm, all secondary to fibrovascular tissue in ectopic locations. In contrast to the more commonly found microphthalmos, children with this complication may present with normal-sized eyes or buphthalmos secondary to the congenital glaucoma. These eyes often begin small and then enlarge.
Persistence of posterior lental fibrovascular tissue can have various appearances within the greater spectrum of PFV. The most commonly recognized, a Mittendorf dot, presents as a small, paracentral retrolental opacity, easily seen with retinoscopy or slit-lamp biomicroscopy. This opacity represents the anterior terminus of the hyaloid artery. When seen with peripheral spoke-like vessels, this finding is often called a “brittle-star” configuration. In the most dramatic form, the entire posterior lens surface may be covered with fibrous tissue as thick as 1 mm.2 This variant must be distinguished from retinoblastoma, complete retinal detachment, retinopathy of prematurity and Coats’ disease, as the management of each entity is unique. In PFV, the vessels are usually regular and may be seen to anastomose. Invasion of the lens with the fibrovascular tissue may cause a lenticular hemorrhage if perfusion persists. Cataract formation can also widely vary from non-existent to partial or complete. Partial cataracts may also progress at any point in life. Persistence of the hyaloid artery may be seen within Cloquet’s canal, connecting the optic nerve to the posterior lens with variable remaining perfusion.
Leukocoria is a common presentation for PFV and can result from a complete pupillary membrane, cataract formation, fibrovascular sheath attachment to the posterior lens capsule or a cloudy cornea secondary to glaucoma (See Figure 1). Children may also first present with strabismus or amblyopia.
A common posterior sign of PFV is the Bergmeister’s papilla emanating from the optic nerve. In isolation, this finding does not typically present visually significant sequelae; however, congenital non-attachment of the retina (also called tent-shaped retinal detachment), which is a more severe form of posterior PFV, has severe visual consequences. Traction is caused by adhesion of the fetal vasculature to the retina, with or without the presence of subretinal fluid. The “brittle-star” sign may be seen at the peak of the tent-shaped detachment and the surrounding spoke-like vessels can be seen well on fluorescein angiography.
Retinal non-attachment is most common in the inferotemporal quadrant, and epiretinal fibrovascular tissue is often found surrounding the area. Macular traction, degeneration and membrane formation may also be found and are often associated with severe visual morbidity. The optic nerve may also be hypoplastic or dysplastic.2,5 Amblyopia may be a presenting symptom in PFV due to a severe but previously undiscovered isolated posterior abnormality without an obvious anterior component.
Diagnosis
The most common means for diagnosis is direct visualization of the persistent vascular remnant. Ultrasonography can be extremely useful to aid diagnosis, especially in the presence of a poor view to the posterior segment. B-scan ultrasonography can assist in ruling out masses and retinal detachments. In addition, CT with contrast will enhance the persistent fibrovascular tissue. Calcifications on B-scan or CT scan should alert the clinician to the possibility of retinoblastoma, as this is a frequent finding that indicates malignancy and is not typically found in PFV. CT scans on children, however, should only be ordered when absolutely necessary due to the possibility of increased malignancy risk.10 MRI is superior in distinguishing soft tissue structures and morphology and does not carry the risk associated with radiation from CT (See Figure 1).5 Fluorescein angiography is another ancillary test that can delineate abnormal vasculature. For example, iridohyaloid vessels appear as radially oriented iris vessels with a hairpin turn around the pupil. Vessels forming a “brittle-star” can also fluoresce.2,5
Prognosis and Treatment
A wide range of treatments and potential outcomes exists for PFV because of the wide spectrum of presentation. Anterior PFV is most often treated with observation, lensectomy and glaucoma management, whether medical or surgical. Posterior PFV is usually associated with a poor visual outcome regardless of intervention due to retinal and optic nerve abnormalities.7,8 Bilateral disease is also typically associated with a poor outcome due to the high prevalence of posterior component.8 Dr. Hunt and colleagues found that eyes without posterior disease often trended toward vision of counting finger or better; this outcome was not statistically significant, however.4 Despite the trend for poor visual outcome, it is possible to obtain useful vision, even with combined anterior and posterior PFV.8 Age at diagnosis is a predictor of final visual outcome, with earlier detection and treatment leading to an increased likelihood of useful vision.5 In one study, post-surgical patients who presented at a mean of 2.4 months achieved a final visual acuity of 20/200 or better, while those that presented at a mean of 4.3 months achieved 20/300 or worse.7 Furthermore, those who received surgical intervention before 77 days of age tended to maintain useful remaining vision.7
|
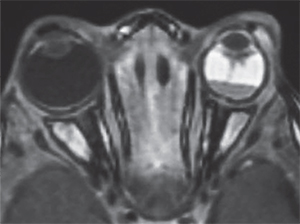
Appropriate patient selection is essential for successful surgical management. Surgery should be avoided in patients with a poor visual prognosis, including severe optic nerve involvement or foveal hypoplasia. Refractive correction and amblyopia therapy are also important for visual rehabilitation, especially in unilateral cases; unfortunately, the density of amblyopia in these cases often makes compliance difficult. Even after surgery, vitreous hemorrhage, glaucoma and rhegmatogenous retinal detachments may recur.8 These potential complications require lifelong monitoring and even in the most ideal situations, visual outcome may be disappointing. All in all, PFV requires meticulous care and follow-up and may involve pediatric ophthalmologists, vitreoretinal surgeons and contact lens specialists. REVIEW
Dr. Farber is a second year resident at SUNY Downstate Medical Center and Dr. Shrier is a retina attending at SUNY Downstate Medical Center.
1. Mary-Sinclair M, et al. Varied manifestations of persistent hyperplastic primary vitreous with graded somatic mosaic deletion of a single gene. Molecular Vision 2014;20:215-230
2. Goldberg, MF. Persistent Fetal Vasculature (PFV): An Integrated Interpretation of Signs and Symptoms Associated with Persistent Hyperplastic Primary Vitreous (PHPV) LIV Edward Jackson Memorial Lecture. Am J Ophthal 1997;124:587-626.
3. Lambert SR, et al. Congenital Fibrovascular Pupillary Membranes: Clinical and Histopathologic Findings. Ophthalmology 2012;119:634-64.
4. Saint-Geniez M, D’Amore P. Development and Pathology of the Hyaloid, Choroidal, and Retinal Vasculature. Int J Dev Biol 2004;48:1045-1058.
5. Pollard Z. Persistent Hyperplastic Primary Vitreous: Diagnosis, Treatment and Results. Tr Am Ophth Soc 1997;95:487-549.
6. Reese A. Persistent Hyperplastic Primary Vitreous. The Jackson Memorial Lecture. Am J Ophthal 1955;40:317-331.
7. Hunt A, et al. Outcomes in persistent hyperplastic primary vitreous. Br J Ophthalmol 2005;89:859-863.
8. Alexandrakis G, et al. Visual Acuity Outcomes with and without Surgery in Patients with Persistent Fetal Vasculature. Ophthalmology 2000;107:1068-1072.
9. Dass A, Trese M. Surgical Results of Persistent Hyperplastic Primary Vitreous. Ophthalmology 1999;106:280-284.
10. Pearce M, et al. Radiation exposure from CT scan in childhood and subsequent risks of leukaemia and brain tumours: A retrospective cohort study. The Lancet 2012;380:499-505.


