The major differential diagnosis of peripheral exudative retinal vascular lesions includes retinopathy of prematurity (ROP), familial exudative vitreoretinopathy (FEVR), Norrie's disease (ND), osteoporosis-pseudoglioma syndrome, Coats' disease and incontinentia pigmenti (IP). In addition, atypical presentations of retinoblastoma, X-linked retinoschisis, or an infectious etiology (i.e. toxocara) must always be considered in these eyes.
Clinical Evaluation
A detailed medical history is critical in the evaluation of a child presenting with a peripheral exudative retinal vascular lesion. The history should include gestational age, weight, length, birth history, neonatal course, and the age when visual loss was first noted. Family history, including known disease in the immediate family or close relatives should be elicited, and available family members should be examined carefully.
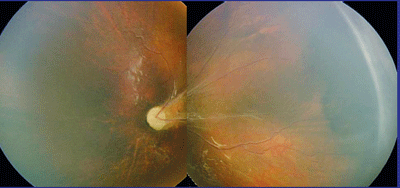 |
| A Retcam montage of the left eye of a patient with autosomal dominant familial exudative vitreoretinopathy. |
Eyes that present with exudative vascular lesions must be carefully examined for cardinal features that help differentiate the various diseases that may present in such a manner. Peripheral retinal capillary nonperfusion, peripheral neovascularization, retinal dragging, intraretinal lipid exudation, subretinal lipid exudation, serous retinal detachment, tractional retinal detachment and vitreous hemorrhage should be noted in detail. Examination under anesthesia may be preferred in children who cannot tolerate a complete retinal exam. We will often obtain fluorescein angiograms in the office, or under anesthesia when necessary. The presence of hearing problems, dermatologic findings, and osteoporosis are key systemic findings that may help to differentiate these diseases.
Disease Features
The diagnosis of ROP can be made based on typical ophthalmoscopic features including the presence of plus disease, peripheral retinal avascularity, vascular proliferation in the form of a ridge or shunt, and typical tractional retinal detachment.1,2 Patients who develop significant ROP almost uniformly have gestational ages of 32 weeks or less and birth weights of 1,500 grams or less. Significant asymmetry and active neovascularization after infancy are unusual in ROP, and a familial history of ROP is rare. These characteristics help to clearly identify ROP as a causative etiology.
The classic finding in FEVR is peripheral retinal capillary nonperfusion. Neovascularization occurs at the border of perfused and nonperfused retina, and tractional retinal detachment, serous exudation and lipid exudation may occur in these regions (See Figure 1). Vitreous hemorrhage and total retinal detachment may occur. While the appearance of ROP and FEVR may be similar, historical features described above for ROP contrast sharply with those of FEVR. Children with FEVR are typically healthy, with normal birth and neonatal courses. Marked asymmetry in disease severity is common in FEVR, but not ROP.
ROP may occasionally resolve spontaneously, while resolution is rare in FEVR. Disease severity in FEVR varies, with approximately 50 percent of patients remaining asymptomatic. These patients are often family members of a patient with advanced disease, who are identified during the screening workup for suspected FEVR. Screening of family members is therefore critical when attempting to diagnose FEVR, as a positive family history will further strengthen a case for the diagnosis.3-6
The osteoporosis pseudoglioma syndrome mirrors FEVR in clinical presentation, but has a unique genetic etiology and, as its name implies, patients will typically demonstrate a concomitant osteoporosis.7
Norrie's disease can mirror the picture of severe FEVR, but is extremely rare, X-linked, symmetrically bilateral and most commonly presents in infancy. Microphthalmia and corneal opacification may occur in Norrie's and, when present, help to differentiate the two disorders. Progressive mental impairment and deafness are important associated features that help solidify the diagnosis.12-14
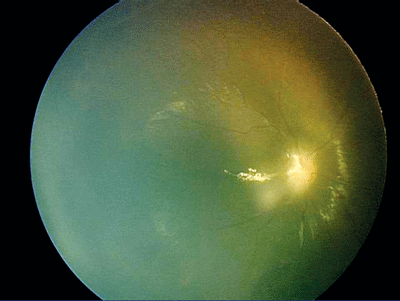 |
| A Retcam photo of the right eye of the patient shown on p. 74. |
Coats' disease is a unilateral, idiopathic retinal vascular abnormality characterized by retinal vascular changes associated with lipid exudation. Most cases are diagnosed before the age of 20 (two-thirds are diagnosed before age 10); the disorder occurs predominantly in boys (85 percent) and is almost always unilateral. Coats' disease presents with varied phenotypic expression, ranging from subtle telangiectatic peripheral retinal vessels to total exudative retinal detachments.8-10 Characteristic features include "light bulb" shaped microaneurysms and capillary non-perfusion on angiography as well as an abundance of lipid exudate.11 The age of presentation, unilaterality, medical history, and typical clinical features help differentiate Coats' from FEVR and ROP.
IP may present with an avascular peripheral retina and neovascularization that can occur either in infancy or later in life. Disease severity is often asymmetric. IP is X-linked and lethal in male patients, and is therefore only seen in female patients. Systemic manifestations are the key to diagnosis, the most reliable of which are the characteristic skin lesions that occur in the neonatal period. These skin lesions are often transient and resolve spontaneously. Other associated manifestations that help support the diagnosis of IP are dental hypoplasia and alopecia.15,16
Molecular Biology and Genetics
The International Incontinentia Pigmenti Consortium demonstrated that mutations in NF-kappaB essential modulator (NEMO) cause incontinentia pigmenti type II.17 The NEMO gene is located on the X-chromosome, and lack of function is usually not compatible with life. Random inactivation of one X chromosome in female patients (lyonization) results in a mosaic; some cells express the normal NEMO gene while others express the mutant form. The NF-kappaB signaling pathway is activated by interleukins, cytokines and other growth factors. Loss of NEMO function prevents normal NF-kappaB activation in response to these factors. (Because of space constraints, refer to a recent review that describes this molecular pathway in greater detail.18)
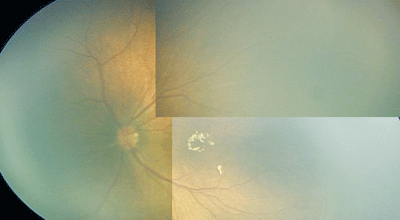 |
| A montage of the left eye of a patient with Incontentia Pigmenti. |
ROP, Norrie's disease, Coats' disease, FEVR, osteoporosis-pseudoglioma syndrome and IP share many features. The primary defect in all of these diseases is the failure of normal vascular development in the peripheral retina. The resulting hypoxia and likely release of angiogenic factors has multiple consequences. The normal regression of the hyaloid vasculature is hindered, neovascularization along the border between perfused and non-perfused retina may develop, and vessels in the border region between perfused and non-perfused retina may display excessive permeability.
Over the last five years, genetic mutations have been identified for each of these diseases. In the cases of Norrie's disease, Coats' disease, FEVR, and osteoporosis pseudoglioma syndrome, the different affected genes all act on the same intercellular signaling pathway. We will first review the elements of this signaling pathway and then discuss the genetic defects for each disease.
Cells must signal between one another to regulate biological processes. One method cells use to communicate is secretion of protein into the extracellular space. The secreted protein is then detected by a neighboring cell via an integral transmembrane receptor protein. The ligand-receptor interaction activates the receptor and triggers an intracellular signaling cascade. Defects in a signaling pathway known as the Wnt pathway underlie Norrie's disease, Coats' disease, FEVR, and osteoporosis pseudoglioma syndrome. This Wnt signaling pathway has been shown to regulate cell adhesion, morphology, proliferation and cell migration in vertebrates. The precise mechanisms by which these genetic mutations cause disease are still being worked out.
It is hypothesized that either a secreted Wnt or norrin (the protein product of the Norrie's disease gene) bind to a cell membrane receptor, which is composed of lipoprotein receptor protein 5(LRP5) and Frizzled-4(FZD4). Activation of the receptor causes up-regulation of intracellular beta-catenin. As beta-catenin accumulates within the cell, it associates at the intracellular side of the membrane with cadherins and can promote cell adhesion and migration. As beta-catenin levels become further elevated, it accumulates within the nucleus and activates transcription in conjunction with co-transcription factors. While the role of this signaling pathway in the vascular development of the peripheral retina is not understood, each of the phenotypically similar diseases discussed arises because of interruptions of this intercellular signaling pathway.
In 2002, researchers demonstrated linkage to 11q13-q23 for autosomal dominant FEVR and refined the disease locus to a genomic region spanning 1.55 Mb.19 The region contained the FZD4 gene, which harbors mutations in affected individuals. Members of the "frizzled" gene family encode 7-transmembrane domain proteins that are receptors for Wnt signaling proteins.
Little is known about the expression pattern of FZD4 in the eye.20 In mice it is expressed during retinal development in the ciliary margin, a region known to contain retinal stem cells. Expression has also been described in Müller glia. In most cases, mutations in FZD4 result in autosomal dominant disease. The mutant protein dimerizes with the normal protein and remains sequestered in the endoplasmic reticulum of the cell, decreasing the amount of receptor in the cell membrane.21 The phenotype is hypothesized to be milder than in Norrie's disease because signaling is attenuated but not absent.
In 2001, another group identified LRP5 mutations in patients with the autosomal recessive osteoporosis-pseudoglioma syndrome.22 They demonstrated that LRP5 and frizzled receptors function together to transduce Wnt signals, placing these two diseases with similar phenotypes into the same genetic pathway. The location of LRP5 within the retina is unknown but presumably overlaps with FZD4, as these proteins must be expressed together to constitute a functional receptor. LRP5 is also expressed in osteoblasts. Loss of function of LRP5 in osteoblasts results in osteoporosis, one of the key clinical features that differentiates osteoporosis-pseudoglioma syndrome from FEVR.23 Pedigrees demonstrating both autosomal dominant and autosomal recessive inheritance have been described.
In 1992, the genetic defect in patients with Norrie's disease was identified. The protein was named norrin, and the researchers deduced from the amino acid sequence that it was a secreted protein.24 Later work placed norrin in a family of genes known as transforming growth factors. Because of the strong clinical similarity between Norrie's disease and FEVR, another group asked whether the secreted norrin protein could act as a ligand for the FZD4/LRP5 receptor.25 This work determined that norrin and FZD4 function as a ligand-receptor pair based on: the similarity in vascular phenotypes caused by norrin and FZD4 mutations in humans and mice; the specificity and high affinity of norrin-FZD4 binding; the high efficiency with which norrin induces FZD4- and LRP5-dependent activation of the classical Wnt pathway; and the signaling defects displayed by disease-associated variants of norrin and FZD4. Because the norrin gene is located on the X chromosome, it most commonly affects males who inherit a defective maternal X chromosome. Norrin is expressed in the outer nuclear, inner nuclear, and ganglion cell layers of the retina as well as the CNS and cochlea.26 This pattern of expression helps explain the deafness and mental retardation that are so frequently associated with this disease.
Coats' disease is characterized by abnormal retinal vascular development and subsequent accumulation of intraretinal and subretinal lipid. The classical form of Coats' disease is almost invariably isolated, unilateral and seen in males. How can this sporadic pattern of disease be explained by a genetic insult? The answer is somatic mutation of a gene on the X chromosome. If cells in the developing retina of a boy lose their only copy of the norrin gene, the retina derived from those mutant cells would develop a region of vascular abnormality. This hypothesis is supported by a female patient with a unilateral variant of Coats' disease who gave birth to a son affected by Norrie's disease. Both carried a missense mutation within the norrin gene on chromosome Xp11.2. Investigators who analyzed the retinas of nine enucleated eyes from males with sporadic Coats' disease were able to demonstrate a somatic mutation in the norrin gene, which was not present within non-retinal tissue.27
The precise mechanism by which mutations in this genetic pathway lead to abnormal vascular development within the peripheral retina is not clear. Inhibition of the Wnt signaling pathway in the retina may directly or indirectly regulate endothelial cells or the scaffold these cells require to form patent vessels. Several laboratories are working to better understand the role of the Wnt pathway in retinal development. Routine genetic testing for mutations in these genes is not currently available.
Manipulation of signaling pathways is proving to be an effective way to treat macular degeneration and diabetic eye disease. These rare diseases have facilitated the identification of another signaling pathway important in retinal vascular development. As researchers begin to unravel how interruption of these signaling pathways leads to abnormal vasculgenesis, perhaps we will learn how to modulate them to our benefit in these and other retinovascular diseases.
Dr. Feiner is in practice at Retina Associates of New Jersey, Teaneck. Dr. Prenner is an assistant clinical professor at Robert Wood Johnson Medical School, UMDNJ. Contact him at UMDNJ Clinical Academic Building, 4th Floor, 125 Patterson St., New Brunswick, NJ 08901. Phone: (732) 235-6333 or email: jonathanprenner@hotmail.com.
1. Committee for the Classification of Retinopathy of Prematurity. An international classification of retinopathy of prematurity. Arch Ophthalmol 1984;102:1130-1134.
2. Cryotherapy for Retinopathy of Prematurity Cooperative Group: Multicenter trial of cryotherapy for retinopathy of prematurity. Snellen acuity and structural outcome at 5 years. Arch Ophthalmol 1996;114:417-424.
3. TasmanW, Augsburger JJ, Shields JA, et al. Familial exudative vitreoretinopathy. Trans Am Ophthalmol Soc 1981;79:211-226.
4. Gitter KA, Rothschild H, Waltman DD, et al: Dominantly inherited peripheral retinal neovascularization. Arch Ophthalmol 1978;96:1601-1605.
5. Slusher MM, Hutton WE. Familial exudative vitreoretinopathy. Am J Ophthalmol 1979;87:152-156.
6. Benson WE: Familial exudative vitreoretinopathy. Trans Am Ophthalmol Soc 1995;93:473-521.
7. Ai M, Heeger S, Bartels CF. Clinical and molecular findings in osteoporosis-pseudoglioma syndrome. Am J Hum Gene 2005;77:741-53.
8. Asdourian G. Vascular anomalies of the retina. In: Peyman GA, Sanders DR, Goldberg MF, eds. Principles and Practices of Ophthalmology, vol. 2. Philadelphia: WB Saunders, 1980.
9. Campbell FP. Coats' disease and congenital vascular retinopathy. Trans Am Ophthalmol Soc 1976;24:365-424.
10. Egerer I, Tasman W, Tomer TL. Coats' disease. Arch Ophthalmol 1974;92:109-112.
11. Gass JDM. Stereoscopic Atlas of Macular Diseases: Diagnosis and Treatment, 3rd edition. St Louis: CV Mosby Co, 1987:384-397.
12. Jacklin HN. Falciform fold, retinal detachment, and Norrie's disease. Am J Ophthalmol 1980;90:76-80.
13. Hausen AC. Norrie's Disease. Am J Ophthyalmol 1968;66:328-332.
14. Townes PL, Roca PD. Norrie's disease (hereditary oculo-acoustic-cerebral degeneration). Am J Ophthalmol 1973;76:797-803.
15. Goldberg, MF, Custis PH. Retinal and other manifestations of incontinentia pigmenti. Ophthalmology 1993;100:1645-1654.
16. Carney RG. Incontinentia pigmenti. A world statistical analysis. Arch Dermatol 1976;112:535-542.
17. The International Incontinentia Pigmenti Consortium : Genomic rearrangement in NEMO impairs NF-kappa-B activation and is a cause of incontinentia pigmenti. Nature 2000;405:466-472.
18. Shastry BS. Recent progress in the genetics of incontinentia pigmenti. J Hum Genet 2000;45:323-6.
19. Robitaille J, MacDonald M, Kaykas A, et al. Mutant frizzled-4 disrupts retinal angiogenesis in familial exudative vitreoretinopathy. Nature Genet 2002;32:326-330.
20. Liu H, Mohamed O, Dufort D, Wallace VA. Characterization of Wnt signaling components and activation of the Wnt canonical pathway in the murine retina. Dev Dyn 2003;227:323-34.
21. Kaykas A, Yang-Snyder J, Heroux M, Shah KV, et al. Mutant Frizzled 4 associated with vitreoretinopathy traps wild-type Frizzled in the endoplasmic reticulum by oligomerization. Nat Cell Biol 2004;6:52-8.
22. Gong Y, Slee R, Fukai N, et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 2001;107:513-523.
23. Westendorf JJ, Kahler RA, Schroeder TM. Wnt signaling in osteoblasts and bone diseases. Gene 2004;341:19-39.
24. Berger W, Meindl A, van de Pol T, et al. Isolation of a candidate gene for Norrie disease by positional cloning. Nature Genet 1992;1:199-203.
25. Xu Q, Wang Y, Dabdoub A, Smallwood PM, et al. Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell 2004;116:883-95.
26. Hartzer M, Cheng M, Liu X, Shastry B. Localization of the Norrie disease gene mRNA by in situ hybridization. Brain Res Bull 1999;49:355-358.
27. Black G, Perveen R, Bonshek R, Cahill M, et al. Coats' disease of the retina caused by somatic mutation in the NDP gene: a role for norrin in retinal angiogenesis. Hum Molec Genet 1999;8:2031-2035.


