Permeability
Vascular permeability, leading to macular edema, causes vision loss through mechanical distortion of the orderly retinal architecture as well as the secondary signal interference. In diabetes, chronic hyperglycemia contributes to alterations in the structural and cellular components of retinal microvasculature. Early on, there is damage to the pericytes responsible for regulating capillary perfusion, which results in microaneurysm formation and impaired regulation of retinal blood flow.1 Damage to endothelial cells responsible for maintaining the blood-retinal barrier allows accumulation of extracellular fluid in the macula. There’s also thickening of the capillary basement membrane and increased deposition of extracellular matrix components.1,2,3 Over time, continued retinal microvasculature damage causes capillary nonperfusion and retinal ischemia, resulting in activation of hypoxia-inducible factor-1 alpha (HIF-1a) via signaling through phosphoinositide 3-kinase and its downstream target, mammalian target of rapamycin (mTOR).4 HIF-1a causes upregulation of VEGF, which mediates vascular permeability.5 Though there are several different VEGF isoforms in the body due to alternative splicing, VEGF-A and placental growth factor mediate vascular permeability and pathologic angiogenesis in the eye. Current agents, ranibizumab (Lucentis, Genentech/Roche), aflibercept (Eylea, Regeneron) and off-label bevacizumab (Avastin, Genentech/Roche), bind VEGF to prevent activation of VEGF receptors, thereby decreasing angiogenesis and vascular permeability, causing regression of diabetic neovascularization and reduction in DME, respectively.2,6
Another target of interest is the Tie-2 receptor tyrosine kinase, which is expressed by endothelial cells. When stimulated, it produces reinforcement of junctional proteins, more interaction with surrounding cells and matrix, and stabilization of the vasculature. Tie-2 is activated by Angiopoietin-1, which results in reduced permeability, whereas Angiopoietin-2 serves as a competitive antagonist that diminishes Tie-2 activity. Ischemic retina results in elevated Ang-2, which is also elevated in patients with DME.7 Following are two investigational agents that target permeability.
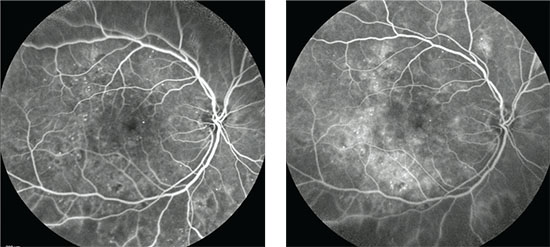 |
| Early (left) and late phase (right) fluorescein angiograms show the typical appearance of an eye with diabetic macular edema. |
• Nesvacumab. Ang-2 antibody Nesvacumab (Regeneron) binds Ang-2, allowing more Ang-1 to activate Tie-2, so that permeability can be diminished. Trials comparing aflibercept alone to aflibercept with Ang-2 antibodies in DME patients are on the horizon.
• AKB-9778. Also affecting the same pathway, AKB-9778 inactivates vascular endothelial-protein tyrosine phosphatase. VE-PTP dephosphorylates Tie-2, causing it to shift to an inactive state. Therefore, AKB-9778 helps keep Tie-2 active by inactivating VE-PTP. This drug is administered by subcutaneous injection, and may serve as an alternative option for patients averse to intravitreal injections. A Phase Ib trial showed that AKB-9778 was well-tolerated, caused significant reductions in retinal thickness and significantly improved visual acuity.8
The Vitreoretinal Interface
For refractory cases of DME, it’s important to assess the vitreoretinal interface carefully, as vitreomacular traction and epiretinal membrane can contribute to macular edema and associated visual loss via tractional forces that exacerbate hyperpermeability. Here’s a look at two approaches addressing interface issues.
• Jetrea (ocriplasmin). The vitreoretinal interface is anchored by extracellular matrix proteins such as laminin, integrins, fibronectin and collagen types VI, VII and XVIII.9 Ocriplasmin, a key ingredient in ThromboGenics’ drug Jetrea, is a recombinant truncated form of human plasmin that has proteolytic activity against these proteins. However, it’s not active against type IV collagen, a component of the internal limiting membrane, thus limiting its toxicity to the retina. When injected into the eye, ocriplasmin can cause pharmacologic vitreolysis and posterior vitreous detachment, offering an alternative to surgery. In one study, ocriplasmin caused resolution of VMT in 26.5 percent of eyes compared to 10.1 percent of placebo eyes within 28 days.10 Ocriplasmin could be used in cases involving VMT with narrow vitreomacular adhesions.
Surgical intervention may be required in cases not amenable to ocriplasmin injection, however, such as those with broad vitreomacular adhesions or epiretinal membrane. In such cases, pars plana vitrectomy with or without membrane peel can serve to release the anteroposterior and tangential tractional forces of the vitreous on the ILM. Gradual restoration of the retinal architecture may result in reduction of edema and improvement in visual acuity. Diabetic eyes may have diffuse residual vitreous cortex after surgical creation of a PVD, which can lead to continued traction and development of ERM.11 Surgical removal of the ILM eliminates the vitreous cortex and any potential proliferative scaffold that could contribute to ERM formation. In eyes with diffuse DME, the ILM becomes thickened and a variety of cells, including myofibroblasts and inflammatory cells, adhere to the inner surface of the ILM, with the potential to further exacerbate edema.11
• Luminate. Another potential treatment targets integrins, which are cell adhesion and cell signaling receptors. The integrin peptide antagonist Luminate (Allegro Ophthalmics) targets receptors involved in both angiogenesis and vitreolysis.
Integrins mediate cell functions, interactions among cells and the extracellular matrix, and activate intracellular pathways that promote angiogenesis. In the body, there are many different types of integrin receptors, but the αvβ3, α5β1, and αvβ5 subtypes are expressed more in diabetic retinopathy.12 Inhibiting α5β1 has been shown to inhibit endothelial cell proliferation and produce regression of choroidal neovascular membranes.13,14 Integrin α3β1 mediates attachment of the vitreous to the retinal surface. Inhibiting α3β1 results in vitreolysis and PVD, which can help patients with VMT, and may serve as an alternative treatment to ocriplasmin. Luminate is currently in Phase II trials for treatment of DME, VMT and AMD.
Inflammation
Inflammation plays an important role in diabetic retinopathy and DME, as leukostasis, prostaglandin upregulation and retinal accumulation of macrophages all occur in diabetes. Diabetics may have abnormally large and rigid leukocytes that adhere to the vascular endothelium, which can contribute to vascular occlusion and ischemia; these leukocytes generate toxic superoxide radicals and proteolytic enzymes.15 The end result is endothelial dysfunction, vascular permeability, retinal nonperfusion and, potentially, angiogenesis, all of which lead to vision loss.
VEGF possesses inflammatory properties through its capacity to mediate microvascular permeability and increase adhesion of leukocytes. VEGF was found to stimulate expression of intracellular adhesion molecule-1 and vascular cell adhesion molecule-1,6 therefore involving the inflammatory cascade, initiating early diabetic retinal leukocyte adhesion and contributing to the development of diabetic vasculopathy. Thus, anti-VEGF agents may also have a therapeutic mechanism that involves limitation of inflammation. Other inflammatory mediators such as tumor necrosis factor-alpha, interleukin-6, interleukin-8, cyclo-oxygenase-2 and monocyte chemotactic protein-1 may also be upregulated in DME.17
• Ozurdex and Iluvien. Recently, there’s been interest in sustained-release corticosteroids, such as the dexamethasone intravitreal implant (Ozurdex, Allergan)18 and the fluocinolone acetonide implant (Iluvien, Alimera),19,20 both of which were approved for the treatment of DME in 2014. Research has suggested that inflammatory cytokines play a larger role in chronic DME than non-chronic DME.18 For this reason, anti-VEGF therapy may not be effective in all patients, because targeting VEGF does not suppress all the inflammatory cytokines involved in chronic DME. Corticosteroids can address this aspect of the pathophysiology by inhibiting the arachidonic acid pathway, resulting in downregulation of prostaglandins, prostacyclin, thromboxanes and leukotrienes. In addition to this anti-inflammatory mechanism, corticosteroids can alter the composition of the endothelial basement membrane by changing the local ratio of two laminin isoforms, suppressing basement membrane dissolution and strengthening tight junctions to limit leakage that can cause macular edema.22,23 In the future, the combination of anti-VEGF therapy and sustained-release corticosteroids is an area of research that may hold promise for treating refractory DME and decreasing the treatment burden of repeated injections.
There are also a couple of new therapies being explored to target the inflammatory component of DME that might serve as alternatives to corticosteroids:
• Infliximab. The drug infliximab inhibits the pro-inflammatory cytokine TNF-alpha, which has been implicated in breakdown of the blood-retinal barrier in diabetic animal models.24 Small studies have yielded inconsistent results in regards to its efficacy in treating refractory DME, however. 25,26 IL-6 is another pro-inflammatory cytokine that may induce expression of VEGF or may directly increase vascular permeability.27,28 IL-6 levels were found to be elevated in patients with DME without PVD, and correlated significantly with severity of disease.29 An IL-6 antibody is currently in development for trials.
• KKS inhibitors. The kallikrein-kinin system is activated in response to vascular injury and leads to a local increase in bradykinin, which further results in pro-inflammatory effects such as vascular permeability, vasodilation and immune cell activation.30 Rodent model studies have shown that diabetes and hypertension increase KKS components in the retina, and inhibition of plasma kallikrein was found to reduce vascular permeability.31 VEGF also leads to increased kallikrein levels in the retina. A KKS inhibitor is being explored for therapeutic efficacy in Phase II trials.
Fibrosis
Retinal fibrosis, which severely distorts and destroys retinal tissue, can result from chronic DME, especially when hard exudates occupy the fovea. Consequently, limiting DME via any of the pathways noted above can minimize fibrosis. As previously mentioned, hypoxia-inducible factor-1a accumulates under hypoxic conditions and serves as a stimulator of VEGF. Hence, there is benefit to inhibiting this pathway that is regulated by the mTOR kinase.4
• Sirolimus. Also known as rapamycin, Pfizer’s Sirolimus is an mTOR inhibitor with the ability to inhibit angiogenesis, permeability, inflammation and fibrosis, making it potentially useful for variety of retinal vascular diseases. It also inhibits protein kinase C and pro-inflammatory mediators such IL-8, endothelial-monocyte activating peptide-II, COX-1 and COX-2.32,33 Rapamycin’s ability to inhibit fibrosis,34 an important contributor to irreversible photoreceptor death and severe visual loss, may fill a void in the currently available DME treatments.
Ischemia
In diabetic retinopathy, enlargement of the foveal avascular zone is detected by fluorescein angiography; it is considered an indication of ischemia and may contribute to macular edema.35 Foveal ischemia in DME causes photoreceptor outer segment shortening and inner segment-outer segment disruption, resulting in outer retinal atrophic changes that correlate with visual loss.36 Unfortunately, there are no treatments available to reverse the atrophic photoreceptor changes associated with macular ischemia.
The pathophysiology of DME is complex and allows for many potential therapeutic targets. While currently laser photocoagulation, anti-VEGF injections and corticosteroids are the most commonly used treatments, there are many intriguing options being explored to address other aspects of DME pathophysiology, including relief of vitreous traction, inflammation and fibrosis. Unfortunately, treatment strategies specifically targeting macular ischemia are lacking, and chronic macular edema can severely limit functional outcomes even if we’re able to reduce macular thickness.37
Dr. Hussain is an ophthalmology resident at the Indiana University School of Medicine. Dr. Ciulla is a volunteer Clinical Professor of Ophthalmology at Indiana University School of Medicine and has an employment relationship with Ophthotech Corporation. He also has been an investigator for Genentech/Roche, Regeneron, Thrombogenics, Allegro, Allergan, Alimera and Pfizer.
1. Ciulla TA, Harris A, Latkany P, et al. Ocular perfusion abnormalities in diabetes. Acta Ophthalmol Scand 2002;80:468.
2. Ciulla TA, Amador AG, Zinman B. Diabetic retinopathy and diabetic macular edema: Pathophysiology, screening, and novel therapies. Diabetes care 2003;26:9:2653-64.
3. Ehrlich R, Harris A, Ciulla TA, Kheradiya N, Winston DM, Wirostko B. Diabetic macular oedema: Physical, physiological and molecular factors contribute to this pathological process. Acta ophthalmol 2010;88:3:279-91.
4. Hudson CC, Liu M, Chiang GG, et al. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol 2002;22:7004.
5. Miller JW, Le Couter J, Strauss EC, Ferrara N. Vascular endothelial growth factor in intraocular vascular disease. Ophthalmology 2013;120:1:106-14.
6. Morello CM. Etiology and natural history of diabetic retinopathy: An overview. Am J Health Syst Pharm 2007;64:S3-7.
7. Hackett SF, Ozaki H, Strauss RW, et al. Angiopoietin 2 expression in the retina: upregulation during physiologic and pathologic neovascularization. J Cell Physiol 2000;184:275-284.
8. Campochiaro PA, Sophie R, Tolentino M, et al. Treatment of diabetic macular edema with an inhibitor of vascular endothelial-protein tyrosine phosphatase that activates Tie2. Ophthalmology 2015;122:3:545-54.
9. Kohno T, Sorgente N, Ishibashi T, Goodnight R, Ryan SJ. Immunofluorescent studies of fibronectin and laminin in the human eye. Invest Ophthalmol Vis Sci 1987;28:506-514.
10. Stalmans P, Benz MS, Gandorfer A, Kampik A, Girach A, Pakola S, Haller JA, Group M-TS. Enzymatic vitreolysis with ocriplasmin for vitreomacular traction and macular holes. N Engl J Med 2012;367:606-615.
11. Kumagai K, Hangai M, Ogino N, Larson E. Effect of Internal Limiting Membrane Peeling on Long-Term Visual Outcomes for Diabetic Macular Edema. Retina 2015;35:7:1422-8.
12. Friedlander M, Theesfeld CL, Sugita M, et al. Involvement of integrins alpha v beta 3 and alpha v beta 5 in ocular neovascular diseases. Proc Natl Acad Sci U S A 1996;93:9764-9769.
13. Ramakrishnan V, Bhaskar V, Law DA, et al. Preclinical evaluation of an anti-alpha5beta1 integrin antibody as a novel anti-angiogenic agent. J Exp Ther Oncol 2006;5:273-286.
14. Umeda N, Kachi S, Akiyama H, et al. Suppression and regression of choroidal neovascularization by systemic administration of an alpha5beta1 integrin antagonist. Mol Pharmacol 2006;69:1820-1828.
15. Miyamoto K, Ogura Y. Pathogenetic potential of leukocytes in diabetic retinopathy. Semin Ophthalmol 1999;14:233-239.
16. Kim I, Moon SO, Kim SH, Kim HJ, Koh YS, Koh GY. Vascular endothelial growth factor expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin through nuclear factor-kappa B activation in endothelial cells. J Biol Chem 2001;276:7614-7620.
17. Hussain RM, Ciulla TA. Treatment strategies for refractory diabetic macular edema: Switching anti-VEGF treatments, adopting corticosteroid-based treatments, and combination therapy. Expert Opin Biol Ther 2016;16:365-374.
18. Boyer DS, Yoon YH, Belfort R, Jr., et al. Three-year, randomized, sham-controlled trial of dexamethasone intravitreal implant in patients with diabetic macular edema. Ophthalmology 2014;121:10:1904-14.
19. Campochiaro PA, Brown DM, Pearson A, et al. Sustained delivery fluocinolone acetonide vitreous inserts provide benefit for at least 3 years in patients with diabetic macular edema. Ophthalmology 2012;119:10:2125-32.
20. Campochiaro PA, Brown DM, Pearson A, et al. Long-term benefit of sustained-delivery fluocinolone acetonide vitreous inserts for diabetic macular edema. Ophthalmology 2011;118:4:626-35 e2.
21. Cunha-Vaz J, Ashton P, Iezzi R, et al. Sustained delivery fluocinolone acetonide vitreous implants: Long-term benefit in patients with chronic diabetic macular edema. Ophthalmology 2014;121:10:1892-903.
22. Tokida Y, Aratani Y, Morita A, Kitagawa Y. Production of two variant laminin forms by endothelial cells and shift of their relative levels by angiostatic steroids. J Biol Chem 1990;265:18123-18129.
23. Stokes CL, Weisz PB, Williams SK, Lauffenburger DA. Inhibition of microvascular endothelial cell migration by beta-cyclodextrin tetradecasulfate and hydrocortisone. Microvasc Res 1990;40:279-284.
24. Joussen AM, Doehmen S, Le ML, et al. TNF-alpha mediated apoptosis plays an important role in the development of early diabetic retinopathy and long-term histopathological alterations. Mol Vis 2009;15:1418-1428.
25. Wu L, Hernandez-Bogantes E, Roca JA, Arevalo JF, Barraza K, Lasave AF. Intravitreal tumor necrosis factor inhibitors in the treatment of refractory diabetic macular edema: a pilot study from the Pan-American Collaborative Retina Study Group. Retina 2011;31:298-303.
26. Sfikakis PP, Grigoropoulos V, Emfietzoglou I, et al. Infliximab for diabetic macular edema refractory to laser photocoagulation: A randomized, double-blind, placebo-controlled, crossover, 32-week study. Diabetes Care 2010;33:1523-1528.
27. Cohen T, Nahari D, Cerem LW, Neufeld G, Levi BZ. Interleukin 6 induces the expression of vascular endothelial growth factor. J Biol Chem 1996;271:736-741.
28. Maruo N, Morita I, Shirao M, Murota S. IL-6 increases endothelial permeability in vitro. Endocrinology 1992;131:710.
29. Funatsu H, Yamashita H, Ikeda T, Mimura T, Eguchi S, Hori S. Vitreous levels of interleukin-6 and vascular endothelial growth factor are related to diabetic macular edema. Ophthalmology 2003;110:9:1690-6.
30. Couture R, Blaes N, Girolami JP. Kinin receptors in vascular biology and pathology. Curr Vasc Pharmacol 2014;12:223-248.
31. Clermont A, Chilcote TJ, Kita T, et al. Plasma kallikrein mediates retinal vascular dysfunction and induces retinal thickening in diabetic rats. Diabetes 2011;60:1590-1598.
32. Rokaw MD, West M, Johnson JP. Rapamycin inhibits protein kinase C activity and stimulates Na+ transport in A6 cells. J Biol Chem 1996;271:32468-32473.
33. Attur MG, Patel R, Thakker G, et al. Differential anti-inflammatory effects of immunosuppressive drugs: Cyclosporin, rapamycin and FK-506 on inducible nitric oxide synthase, nitric oxide, cyclooxygenase-2 and PGE2 production. Inflam Res 2000;49:1:20-6.
34. Salas-Prato M, Assalian A, Mehdi AZ, Duperre J, Thompson P, Brazeau P. Inhibition by rapamycin of PDGF- and bFGF-induced human tenon fibroblast proliferation in vitro. J Glaucoma 1996;5:54-59.
35. Sakata K, Funatsu H, Harino S, Noma H, Hori S. Relationship between macular microcirculation and progression of diabetic macular edema. Ophthalmology 2006;113:8:1385-91.
36. Lee DH, Kim JT, Jung DW, Joe SG, Yoon YH. The relationship between foveal ischemia and spectral-domain optical coherence tomography findings in ischemic diabetic macular edema. Invest Ophthalmol Vis Sci 2013;54:1080-1085.
37. Douvali M, Chatziralli IP, Theodossiadis PG, Chatzistefanou KI, Giannakaki E, Rouvas AA. Effect of macular ischemia on intravitreal ranibizumab treatment for diabetic macular edema. Ophthalmologica 2014;232:136-143.


