Ciera Maffei and Amanda J. Howe,
The efficacy of ophthalmic drugs depends not only on their mechanisms of action and formulation, but also on their ability to reach the target tissues and to maintain their desired effects once they get there (pharmacokinetics). In ophthalmology, the vast majority of treatments for anterior eye disease, such as dry eye, allergy and microbial keratitis, take the form of topical ophthalmic solutions. This mode of delivery is generally preferable to systemic routes in terms of localized results and side-effect profiles, but the innate barriers of the ocular surface microenvironment make drug penetration and residency challenging. Here's what you need to know about challenges to anterior segment drug delivery, as well as the advantages of ocular anatomy and physiology.
Natural Barriers
When an eye drop is instilled into the tear film, it's immediately diluted by natural lacrimation. The tear film and blinking action are designed to eliminate foreign particles, so much of the drug that's initially applied to the eye is lost to spillage onto the skin and drainage through the nasolacrimal duct.1,2 These innate defense mechanisms, coupled with the cul-de-sac's tendency to take up normal volumes of approximately 7 to 10 µL of fluid, and normal tear turnover rates ranging from 0.5 to 2.2 µL/minute (often estimated at roughly 16 percent per minute), are impressive when combating infection but limit typical contact time of the drug with ocular tissues to a sub-optimal one to two minutes.1,3,4 Furthermore, during this window, only about 5 percent of administered drug will enter the eye, and reflex tearing may compound the situation by diluting the tear fluid even further.5
Following drop instillation, the peak concentration is usually reached after 20 to 30 minutes, but this is typically two orders of magnitude lower than the administered dose, even for lipophilic compounds.6 Lipophilic drugs are cleared by aqueous humor turnover, at a rate of about 3 µL per minute, or eliminated by uveal blood flow at about 20 to 30 µL per minute. A drug can only enter the uveal blood flow, however, if it can penetrate the corneal endothelium.6 Surprisingly, distribution to the lens is much slower than that to the uvea, but this is due to their distinct natures: the lens is tightly packed and protein-rich whereas the uvea is porous.6
Corneal Entry
At the tear film-epithelial interface, the drug can follow one of two paths: the corneal or so-called non-corneal (stromal and conjunctival) routes of entry. In the tear film, the drug reaches its first series of barriers to corneal penetration: the corneal epithelium, which is lipophilic and consists of stratified squamous cells five to seven cell layers thick. The epithelium forms a barrier to the outside environment and also regulates the passage of fluid through paracellular pathways. The leakage of this fluid controls corneal thickness and transparency as well as corneal nutrition. However, tight junctions positioned at the apical points of the most superficial cells must have low resistance to permit nutrient passage.7
Immediately posterior to the corneal epithelium, and in contact with the basal columnar cells, is tough, collagenous Bowman's membrane. The hydrophilic, lamellar stroma, posterior to Bowman's membrane, further complicates entry and accounts for 90 percent of corneal thickness. Since it's hydrophilic, the stroma is a strong barrier to lipophilic molecules, though it lacks tight-junction complexes.8 Past the stroma lies the single-cell layer of Descemet's membrane and the extracellular matrix secreted by the deepest layer of the cornea, the endothelium. The corneal endothelium is a single cuboidal cell layer in thickness, and is a lipophilic region responsible for permitting leakage of nutrients from the aqueous humor to the cornea and for transporting water from the avascular cornea9 into the anterior chamber. Thus, the lipophilic-hydrophilic-lipophilic nature of the cornea, as illustrated, is a major contributor to the challenge of drug permeability.
Non-Corneal Entry
The conjunctiva is composed of the surface epithelium and substantia propria. The stratified, non-keratinized conjunctival epithelium contains goblet cells that act as primary mucin producers. The substantia propria is a layer of connective tissue that contains an abundance of immune cells, and it's believed to actually consist of two layers itself: a deep, fibrous layer and superficial lymphoid layer. The conjunctiva has approximately 17 times the surface area of the cornea and is generally "leakier," making drug permeability easier in the conjunctiva than the cornea.10,11
The conjunctival epithelium is stratified squamous cells at the lids (where the conjunctiva ranges from five to six cell layers) and transitions to stratified columnar epithelium toward the cornea (10 to 15 cell-layers thick).11 High levels of vascularization throughout the conjunctiva are another differentiating factor when compared to the cornea, as this vascularization contributes to some level of drug loss to systemic circulation.11
As we discussed in last month's column, tight junctions lie between conjunctival epithelial cells and are composed of four types of proteins: occludins; claudins; junction adhesion molecules; and tricellulin. Tight junctions create a sealed barrier to prevent the entry of microorganisms, pollutants, allergens and the like into ocular tissues. Not only does the barrier prevent trespassers' access, it also inhibits drug passage via the paracellular route. Tight junction permeability depends on several factors including maturity of epithelia, environmental conditions and chemical factors (e.g., drugs, vitamins, hormones and enzymes).4 Calculated pharmacologic modulation of these junctions represents an opportunity for treatment, as drug delivery profiles may be augmented via controlled "loosening" of the junctions; conversely, enhanced barrier protection may be achieved if a drug is capable of bolstering tight junctions.
Beneath the conjunctiva is the white, collagenous layer of the sclera. The extracellular matrix is composed of three types of fiber: collagen; re--ticular and elastic. Collagen fibers, the largest of the three types, consist of thin fibrils tightly compacted to form cord- or tape-like shapes.12 Reticular fibers are more delicate. These fine fibrils can be individualized or grouped but are continuous with collagen fibers.12 The elastic fibers are the smallest and aren't typically packed together. While the collagen fibrillar system forms the scaffold for tissues and cells, the elastin system acts as a buoy to homogeneously distribute stress and establish tissue resilience.12 Therefore, the collagenous sclera is responsible for maintaining the eye's shape by resisting intraocular pressure, and it constitutes approximately 80 percent of the surface area of the globe.11
The sclera is structurally similar to the corneal stroma and displays comparable small-molecule permeability as well. The sclera is generally poorly vascularized and large-molecule transport through the sclera may occur through perivascular spaces or as diffusion between scleral fibrils.
Natural Reservoirs
Some features of ocular anatomy actually provide natural reservoirs, or absorption/adsorption mechanisms for internal "sustained-release." The cells of corneal epithelium itself, like other stratified epithelia throughout the human body, turn over in approximately five to seven days.9 Since the half-life of an epithelial cell has been shown to be around three days, drug-binding to the epithelial cell is one of these potential internal sustained-release mechanisms.13
The U.S. Food and Drug Administration requires that toxicity studies for ophthalmic drugs be carried out in pigmented rabbits, or that a melanin toxicity study be done in their place, which highlights the important role of pigment in pharmacokinetics. Melanin binding affects the metabolism and clearance of drugs, so albino rabbits no longer suffice as appropriate subjects for these studies.
This topic has been of interest since the first publication on the binding of phenothiazines to ocular melanin in 1962.14 Subsequently, many studies have explored the possible effects of drug accumulation in pigmented tissues but particular attention has been paid to ocular toxicity. Contrasting views have been published, with one study in particular asserting that "the binding of drugs to eye melanin is not predictive of ocular toxicity,"15 but the consensus seems to be that they are indeed related.
Once topically administered drugs enter the tear film and enjoy their brief contact time with the ocular surface, only lipophilic compounds remain in the epithelium.6 From there, they're slowly released into the stroma and anterior chamber. They may rest in the aqueous humor, but the drug can also access the iris and ciliary body where it has the opportunity to bind to melanin. Once bound, it may form a reservoir and thereby be gradually released into surrounding cells, prolonging drug activity.6
Melanin content may also affect transscleral delivery of drug to the retina. In one study, a 3-mg suspension of celecoxib was injected periocularly into albino and pigmented rats.16 The concentrations of melanin were 200 ±30 µg/mg tissue in the choroid-RPE, 12 ±4 in the sclera, and 3 ±0.2 in the retina of pigmented rats. In albino rats, melanin was not detected in any tissue except the choroid-RPE, where melanin-like activity was a hundredfold less than in pigmented rats.16 The retinal (p=0.001) and vitreal (p=0.001) absolute bioavailability of celecoxib in the treated eyes was approximately 1.5 times higher in albino than in pigmented rats, suggesting significant binding of celecoxib to melanin, and that its accumulation/retention in the melanin-rich choroid-RPE of pigmented rats hinders transscleral drug availability. The authors also assert that this phenomenon is true of sustained-release microparticle systems.16
Vitreous
The idea of sustained release gained ground in the 1970s, and has continued to do so. The intent is to deliver a localized concentration of drug over the entire diseased area for an optimal duration while avoiding the adverse effects of peak/trough kinetics seen with injections.17 One of the most challenging therapeutic targets is the macula, but innovative use of the vitreous is one avenue being explored.
Because, as we noted previously, topical drugs need to be administered frequently just to maintain therapeutic levels in the anterior segment, they're not suitable for posterior drug delivery.17 Furthermore, the adverse ef-fects of intravitreal injections, in--cluding endophthalmitis, vitreous hem-orrhage and cataract, underscore the need for novel delivery methods.17
Currently under investigation is trans-scleral delivery, which is less invasive and suffers from minimal systemic absorption. Iontophoretic transfer of charged drugs into tissues using electrical currents is one method, but sustained release systems seem to be better accepted.
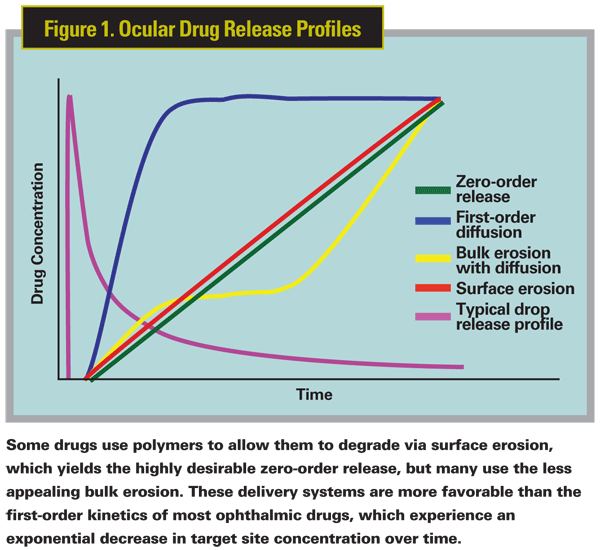
In the years since Vitrasert and Pilosert came off the market, strategic developments have been made in ophthalmic drug delivery systems. These newer systems, including Ocusert (Merck) and Lacrisert (Aton Pharma), are specifically designed and adapted to the target tissue, the physio-chemical properties of the active drug compound and the desired kinetics of ocular release. Although Vitrasert exhibited nearly zero-order kinetics, a system in which the bio-availability of an administered drug remains constant throughout the delivery period, its major drawback was that it was non-biodegradeable and required surgical implantation and removal after it emptied. Newer delivery systems have combined the drug with various polymeric compounds to create matrix and reservoir biodegradable delivery systems. The erosion rates and degradation of these polymers can be modulated to allow for the desired ocular kinetics of drug release. The polymers that degrade via surface erosion give drugs the highly desirable zero-order release, but many use bulk erosion which doesn't behave optimally. These controlled delivery systems are naturally more favorable than the first-order kinetics of most ophthalmic drugs, which experience an exponential decrease in target site concentration over time and, thus, require more frequent dosing. Figure 1 illustrates these principles.
Even without this engineering, the vitreous can act as a compartment for sustained release, and particulate substances may be suspended there.18
Ocular anatomy and physiology provide many opportunities for both inhibition and enhancement of drug delivery. Certain drugs, such as glaucoma treatments, demonstrate the need to penetrate ocular tissues to gain entry into the anterior chamber, but others, like anti-allergics, are most effective if they maintain high concentrations within the surface tissues. Still other drugs (e.g., anti-inflammatories) demonstrate efficacy in both situations, depending on the disease target. Drug delivery techniques include potential avenues to overcome anatomical barriers as well as strategies that take advantage of other properties of the ocular surface.
Dr. Abelson, an associate clinical professor of ophthalmology at
1. Chun DK, Shapiro A, Abelson MB. Ocular Pharmacokinetics. In: Albert DM, Jakobiec FA, eds. Principles and Practice of Ophthalmology, v. 1, 3rd ed.
2. Maurice DM. The dynamics and drainage of tears. Int Ophthalmol Clin 1973;13:1:103-16.
3. Mishima S, Gasset A,
4. Kaur IP, Smitha R. Penetration enhancers and ocular bioadhesives: Two new avenues for ophthalmic drug delivery. Drug
5. Gaudana R, Jwala J, Boddu SH,
6. Urtti A. Challenges and obstacles of ocular pharmacokinetics and drug delivery. Adv Drug Deliv Rev 2006;58:11:1131-5.
7. Stiemke MM, McCartney MD, Cantu-Crouch D, Edelhauser HF. Maturation of the corneal endothelial tight junction. Invest Ophthalmol Vis Sci 1991;32:10:2757.
8. Macha S,
9. Gipson IK, Joyce NC. Anatomy and cell biology of the cornea, superficial limbus, and conjunctiva. In: Albert DM, Jakobiec FA, eds. Principles and Practice of Ophthalmology, v. 1, 3rd ed.
10. Ashton P, Podder SK, Lee VH. Formulation influence on conjunctival penetration of four beta blockers in the pigmented rabbit: A comparison with corneal penetration. Pharm Res 1991;8:9:1166-74.
11. Ahmed I. The Noncorneal route in ocular drug delivery. In:
12. Kligman AM, Zheng P,
13. Cenedella RJ, Fleschner CR. Kinetics of corneal epithelium turnover in vivo. Studies of lovastatin. Invest Ophthalmol Vis Sci 1990;31:10:1957-62.
14. Potts AM. Uveal pigment and phenothiazine compounds. Trans Am Ophthalmol Soc 1962;60:517-52.
15. Leblanc B, Jezequel S, Davies T, et al. Binding of drugs to eye melanin is not predictive of ocular toxicity. Regul Toxicol Pharmacol 1998;28:2:124-32.
16. Cheruvu NP, Amrite AC, Kompella UB. Effect of eye pigmentation on transscleral drug delivery. Invest Ophthalmol Vis Sci 2008;49:1:333-41.
17. Booth BA, Vidal Denham L, Bouhanik S, et al. Sustained-release ophthalmic drug delivery systems for treatment of macular disorders: Present and future applications. Drugs Aging 2007;24:7:581-602.


