Diagnosis, Workup and Treatment
A key initial consideration was whether the patient’s abduction deficit was isolated, or rather, one component of a more systemic etiology that could encompass the patient’s additional complaints (e.g., dizziness, decreased hearing). Given the lack of overt vasculopathic risk factors, a broad differential diagnosis for abduction deficit was considered.
Magnetic resonance imaging of the brain with venography (MRI/MRV) was completed on admission, which returned negative for cerebrovascular accident, sinus thrombosis or mass, but was notable for enhancement in the right mastoid air cells with extension to the petrous apex (See Figure 1).
Lumbar puncture was performed to rule out potential infectious or autoimmune disease, which revealed normal opening pressure, protein count of 31 mg/dL, red cell count of 1, white cell count of 0, and glucose of 62 mg/dL. Laboratory investigation was concurrently initiated, with studies for potential etiologic factors such as myasthenia gravis (acetylcholine receptor antibodies) and thyroid disease (thyroid stimulating hormone, free T4), all returning within normal limits. Complete blood count, metabolic panel, erythrocyte sedimentation rate, and C-reactive protein were similarly within normal limits.
|
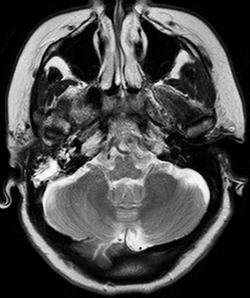
Given the imaging findings, one particular diagnosis was considered. The MRI finding of petrous apex disease ipsilateral to the abduction deficit raised the question of Gradenigo’s syndrome, classically characterized by petrous apex inflammation, sixth nerve palsy, and orbitofacial pain (involvement of cranial nerve five) in the setting of otitis media. The etiology of the sixth nerve palsy is understood anatomically, given the course of the sixth nerve as it passes the petroclinoid ligament. As such, the Otolaryngology Service was consulted. Otoscopic exam findings were consistent with a clear serous effusion, accounting for the patient’s complaint of decreased hearing, with no mastoiditis or otitis media present. The MRI findings were considered post-infectious changes secondary to the patient’s recent episode of bronchitis. Thus, Gradenigo’s syndrome was thought not to be the cause of the patient’s abduction deficit in this case.
Attention returned to the physical exam for further information. A thorough neurologic exam was completed by the Wills Eye Neuro-ophthalmology Service. Speech was clear and fluent, with sensation and motor strength preserved. Gait was normal but the patient was observed to walk with short steps. However, the exam was notable for dysmetria on finger-to-nose testing with areflexia at the ankles, worse than documented on exam at admission.
The findings of ataxia and areflexia on exam were suggestive of Miller Fisher syndrome. Her history of recent viral illness (bronchitis) was consistent; however, lumbar puncture with normal cerebrospinal fluid (CSF) protein level was not. Options for auxiliary diagnostic testing were considered, and the decision was made to repeat the lumbar puncture. Subsequent fluid studies on repeat puncture were significant for an elevated CSF protein of 516 mg/dL, white cell count of 0, red cell count of 1, and glucose of 72 mg/dL, only five days after the initial lumbar puncture was completed.
In concert with the Neurology Service, the diagnosis of Miller Fisher syndrome was made, with all clinical features of ophthalmoplegia, ataxia and areflexia present and CSF studies supportive. The patient was treated with intravenous immunoglobulin (IVIG), tolerating a five-day treatment course well, with subsequent discharge to home. At eight weeks follow-up, the patient’s symptoms were entirely resolved with no residual abduction deficit, ataxia or areflexia on exam.
Discussion
Miller Fisher syndrome is an uncommon variant of Guillain-Barré syndrome characterized by ophthalmoplegia, ataxia and areflexia.2 As with other variants of Guillain-Barré syndrome, the condition is described as an immune-mediated polyneuropathy preceded by a viral illness, commonly Campylobacter jejuni or Haemophilus influenza, amongst others. Of all cases of Guillain-Barré syndrome in the United States, the Miller Fisher variant comprises 5 percent of the total, with an estimated incidence of 0.09 in 100,000.3
While largely a clinical diagnosis when all features (e.g., ophthalmoplegia, areflexia and ataxia) are present, auxiliary testing is available. These modalities include CSF fluid studies, serologic testing, electromyography and imaging.
CSF fluid studies are a common option, classically characterized by the albumino-cytologic dissociation (elevated CSF protein in the setting of an otherwise normal cell count) typical for Guillain-Barré spectrum disease. This case demonstrated an important pearl regarding the use of CSF fluid analysis in the diagnosis of Guillain-Barré spectrum disease. Up to 20 percent of patients will not demonstrate CSF abnormalities at one week after clinical onset of disease; thus, a “negative” CSF fluid analysis early in the disease course does not rule out Miller Fisher syndrome. It was for this reason that a repeat lumbar puncture was considered in this case, and ultimately proved useful diagnostically.
The advent of serologic testing for Miller Fisher syndrome, namely the anti-GQ1b antibody, is of particular importance. Pathogenesis in Guillain-Barré syndrome is thought to be due to molecular mimicry between an antigen present in the preceding viral illness and an endogenous host protein. The ensuing humoral immune response thus targets both the viral and host protein in question. In the case of Miller Fisher syndrome, the endogenous protein is thought to be the ganglioside GQ1b, a cellular component found abundantly in the oculomotor nerves, thus explaining ophthalmoplegia as a clinical feature.3
| ||||||||||||||||
Additional diagnostic studies may also be utilized. Electromyography reveals reduced sensory nerve action potentials with absence of H reflexes.7 Imaging findings are generally nonspecific, though individual case reports have described enhancement of cranial nerves in cases of Miller Fisher syndrome.8 Treatment includes modalities aimed at reducing levels of circulating IgG antibodies. IVIG and plasmapheresis are traditional treatment approaches, with both options found equally efficacious in meta-analysis review.9
This case demonstrates the important role of the neurologic exam in cases of abduction deficit where systemic involvement is suspected. Without objective documentation of areflexia and ataxia, Miller Fisher syndrome cannot be diagnosed clinically. The consideration of alternative diagnoses, namely Gradenigo’s syndrome, was also notable. While Gradenigo’s syndrome is now rare, typically seen with untreated cases of severe otitis media in children, MRI findings consistent with petrous apex disease necessitate further evaluation. As such, clinical correlation in concert with the Otolaryngology Service was required in this case.
In summary, ophthalmoplegia in the setting of areflexia or ataxia on neurologic exam should raise suspicion for Miller Fisher syndrome, a rare clinical variant of Guillain-Barré syndrome. CSF fluid studies and serum titers of the anti-GQ1b antibody are particularly useful in confirming the diagnosis. REVIEW
The author would like to thank Mark Moster, MD, of the Wills Eye Institute Neuro-ophthalmology Service, for his time and assistance.
1. Motamed, M and Kalan A. Gradenigo’s syndrome. Postgrad Med J 2000;76:559-60.
2. Fisher M. An unusual variant of acute idiopathic polyneuritis (syndrome of ophthalmoplegia, ataxia and areflexia). N Engl J Med 1956;255:57–65.
3. Lo YL. Clinical and immunological spectrum of the Miller Fisher syndrome. Muscle Nerve 2007;36:615–627.
4. Arányi Z, Kovacs T, Sipos I, et al. Miller Fisher syndrome: Brief overview and update with a focus on electrophysiological findings. Eur J Neurol 2012;19:15-20, e1-3.
5. Nishimoto Y, Odaka M, Hirata K, et al. Usefulness of anti-GQ1b IgG antibody Testing in Fisher syndrome compared with cerebrospinal fluid examination. J Neuroimmunol 2004;148:200-5.
6. Koga M, Gilbert M, Takahask M, et al. GQ1b-seronegative Fisher syndrome: Clinical features and new serological markers J Neurol 2012. [Epub ahead of print]. http://www.ncbi.nlm.nih.gov/pubmed/22218648.
7. Arányi Z, Kovacs T, Sipos I, et al. Miller Fisher syndrome: Brief overview and update with a focus on electrophysiological findings. Eur J Neurol 2012;19:15-20, e1-3.
8. Garcia-Rivera CA, Rozen TD, Zhou D, et al. Miller Fisher syndrome: MRI Findings. Neurology 2001;57:1755.
9. Hughes RAC, Swan AV, van Doorn PA . Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database of Systematic Reviews 2010, Issue 6. Art. No.: CD002063. DOI: 10.1002/14651858.CD002063.pub4.


