Presentation
A 33-year-old Caucasian male presented to the Wills Eye Emergency Room after noticing blurry vision temporally in his right eye for three days. He denied trauma, new floaters, pain or headache but did see one flash of light.
Medical History
The patient's past medical history was significant for recently diagnosed hypertension. He had started ramipril the day before his symptoms began. He did not have a known diagnosis of demyelinating disease or an inflammatory condition. He had no past surgical history. The patient denied tobacco, alcohol or illicit drug use. His family history was noncontributory. His review of systems was significant for a flu-like illness, which had recently resolved.
Examination
Best-corrected visual acuity was 20/20 in the right eye and 20/20 in the left eye. There was a questionable slight relative afferent pupillary defect in the right eye. The patient was not proptotic, and there was no resistance to retropulsion or obvious mass. Color plates were a brisk 10/10 in both eyes; however, the patient reported subjective bright desaturation in the right eye. Amsler grid testing also revealed a relative scotoma supero-temporally in the right eye. Intraocular pressures and anterior segment examinations were normal. Dilated fundus examination of the right eye was significant for a hyperemic optic disc with mildly blurred margins (See Figure 1). The posterior exam of the left eye was unremarkable. The patient was sent for an MRI of the brain and orbits with and without gadolinium. Basic metabolic panel, complete blood count with differential, ACE, RPR and ANA were drawn. MRI showed no abnormal enhancement of the optic nerves, no mass and no white matter lesions. All lab work was within normal limits as well.
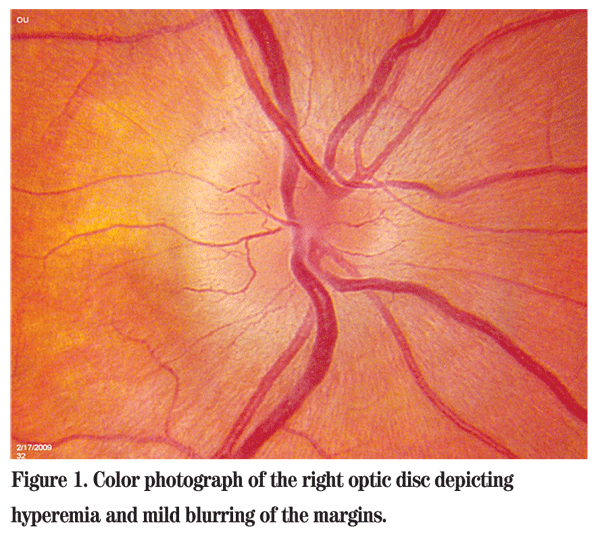
Diagnosis, Workup and Treatment
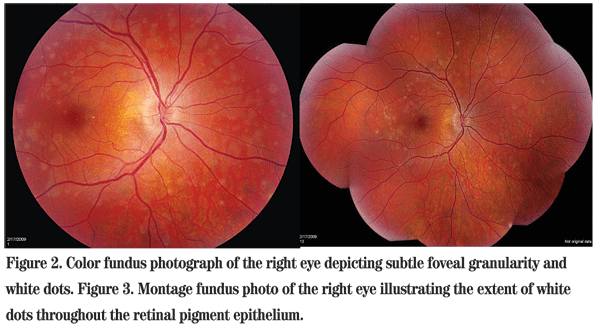
The patient was discharged from the emergency room and referred to the Wills Eye Neuro-Ophthalmology Service three days later. At that time, he reported a subtle worsening of his symptoms. Refraction revealed mild myopia in both eyes. Repeat fundus exam showed subtle foveal granularity in the right eye. In addition, white dots at the level of the retinal pigment epithelium were scattered throughout the posterior pole (See Figures 2 & 3). There was no associated uveitis.
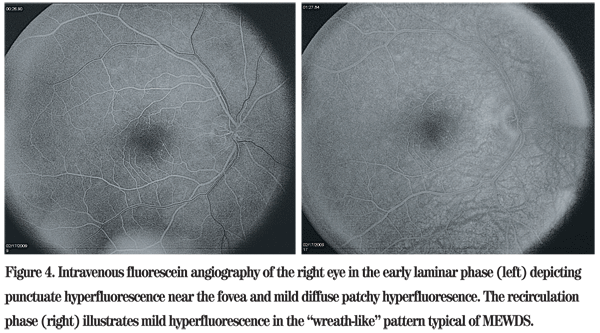
The differential diagnosis of mild optic disc edema and multiple white dots in a 33-year-old myopic Caucasian male includes multiple evanescent white dot syndrome (MEWDS), acute posterior multifocal placoid pigment epitheliopathy (APMPPE), serpiginous choroidopathy, birdshot retinochoroidopathy, multifocal choroiditis and pan-uveitis syndrome, punctuate inner choroidopathy, acute macular neuroretinitis and acute zonal occult outer retinopathy. An enlarged blind spot is not a typical finding in APMPPE, but can be seen in all of the other conditions mentioned. Subtle foveal granularity noted on clinical examination raised suspicion for MEWDS, particularly in a young myope with acute unilateral disease and a history of photopsia. Intravenous fluorescein angiography was subsequently performed, which illustrated hyperfluorescent spots with late staining, confirming the diagnosis.
We discussed the diagnosis with the patient, who elected to monitor his vision with an Amsler grid and return regularly for retinal examination.
Five months later the patient returned for follow-up and reported resolution of his symptoms. Visual acuity was 20/20 in both eyes and a 24-2 Humphrey visual field of the right eye had returned to normal. Dilated fundus exam of the right eye revealed a few scattered white dots, some residual retinal pigment epithelial atrophy and resolution of disc edema with no pallor.
Discussion
Multiple evanescent white dot syndrome is an idiopathic condition whose pathogenesis is poorly understood, but appears to involve inflammation of the neurosensory retina, retinal pigment epithelium and choroid. Afflicted patients develop unilateral acute loss of vision, relative scotomas and photopsias. Examination reveals white dots scattered throughout the posterior pole, usually hugging the vascular arcades and sparing the fovea. The lesions are at the level of the retinal pigment epithelium, choroid or deep retina. Granularity of the fovea, subtle disc edema, mild vitritis, dyschromatopsia, retinal vascular sheathing and afferent pupillary defects have also been reported.
Approximately 80 percent of MEWDS cases occur in women, with an average age of 27 years and a range of 14 to 47 years. In one study, about half of all affected patients reported a viral illness preceding onset of visual symptoms, which suggests either an infectious or autoimmune etiology. Immunohistochemistry has not yet illustrated antibodies bound to retinal antigens, despite the finding of increased serum immunoglobulins in MEWDS patients.
Visual field defects are often more extensive than clinical appearances suggest. The most common defect is enlargement of the blind spot, but MEWDS can also cause central, cecocentral and arcuate scotomas.
Fluoroscein angiography illustrates hyperfluorescence with late leakage in areas corresponding to the white funduscopic lesions. Additional lesions that are not clinically evident can be observed angiographically, as well. Indocyanine green angiography classically demonstrates hypofluorescent lesions, and patients with enlarged blind spots have hypofluorescent rings surrounding the optic disc. Reduced ERG potentials indicative of photoreceptor dysfunction also help identify MEWDS.
Recent optical coherence tomography analysis of MEWDS lesions has shown accumulation of an unknown substance in the subretinal space with disruption of retinal cones. The material largely regresses when clinical symptoms resolve, but some OCT evidence of lesions remains for several months afterwards.
No effective treatment currently exists for MEWDS, but the majority of patients fully recover within 10 weeks. Some may have persistent field defects, and occasionally a patient will experience recurrences. Systemic immunosuppression with cyclosporine may help reduce recurrence rates in such instances.
The author thanks Jennifer Hall, MD, Wills Eye Institute Department of Neuro-Ophthalmology, and Jeremy Wolfe, MD, Wills Eye Institute retina fellow, for their time and assistance.
1. Jampol LM, Sieving PA, Pugh D, Fishman GA, Gilbert H. Multiple evanescent white dot syndrome. I. Clinical findings. Arch Ophthalmol 1984;102:671-4.
2. Jampol LM, Tsai L. Multiple evanescent white dot syndrome. In: Ryan SJ, ed. Retina.
3. Mamalis N, Daily MJ. Multiple evanescent white-dot syndrome. A report of eight cases. Ophthalmology 1987;94:1209-12.
4. Slusher MM, Weaver RG. Multiple evanescent white dot syndrome. Retina 1988;8:132-5.
5. Sieving PA,
6. Huang J, Spaide R. Appearance of brown areas after resolution of the acute phase of multiple evanescent white dot syndrome. Retina 2004;24:814-6.
7. Gross NE, Yannuzzi LA, Freund KB, Spaide RF, Amato GP, Sigal R. Multiple evanescent white dot syndrome. Arch Ophthalmol 2006;124:493-500.
8. Aaberg TM, Campo RV, Joffe L. Recurrences and bilaterality in the multiple evanescent white-dot syndrome. Am J Ophthalmol 1985;100:29-37.
9. Hall JK, Volpe NJ. Autoimmune Retinopathies. In: Lorenz B, Borruat F-X. Pediatric Ophthalmology, Neuro-Ophthalmology, Genetics. In: Krieglstein GK, Weinreb RN: Essentials in Ophthalmology. Springer-Verlag 2008, vol 7:163-180.


