 |
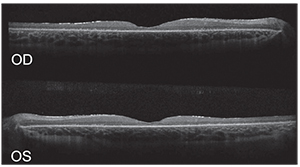
| Figure 2. Spectral-domain optical coherence tomography of the right and left eyes showing an abnormal foveal contour with retinal thinning and photoreceptor loss. Large intraretinal cystic spaces were noted, mainly within the left eye. |
Given our patient’s history and exam, a differential diagnosis was directed toward retinal dystrophies that could cause decreased visual acuity and nyctalopia as well as pigmentary changes in the macula and mid-periphery.
Further workup was directed at identifying the nature of his apparent retinal dystrophy. OCT showed an abnormal foveal contour with macular thinning and photoreceptor loss in both eyes (See Figure 2).
Additionally, macular intraretinal cystoid spaces were noted, largely in the left eye. Both eyes showed surface gliosis. Fundus autofluorescence was notable for mottled hypofluorescent changes throughout the macula surrounding a relatively normal fovea (See Figure 3). Electrophysiology testing revealed near isoelectric responses on multifocal and full-field electroretinogram of each eye. Given the aforementioned non-specific ophthalmic findings, genetic testing with a retinal dystrophy panel was performed at the initial visit. Due to the presence of intraretinal cystoid spaces, the patient was started on dorzolamide ophthalmic drops twice daily in both eyes.
At his three-month follow-up visit, fundus examination was unchanged from the previous visit. Repeat OCT showed resolution of cysts (See Figure 4). Despite these findings, best-corrected visual acuity was now 20/40 in each eye and the parents reported a notable decrease in visual function. Genetic testing revealed a well-known homozygous 1.02kb deletion in exon 7 to 8 of the CLN3 gene.
Based on these examination and genetic test findings, a diagnosis of juvenile neuronal ceroid lipofuscinosis was made.
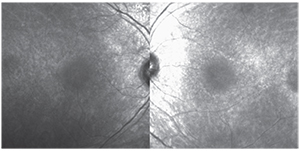 |
| Figure 3. Fundus autofluorescence of the right and left eyes depicting mottled hypofluorescent changes throughout the macula surrounding a relatively normal fovea. |
 |
| Figure 4. Spectral-domain OCT of the right and left eyes at follow-up visit show resolution of cystoid macular changes, with persistence of retinal thinning and abnormal foveal contour. |
The neuronal ceroid lipofuscinoses are a group of inherited progressive neurodegenerative disorders, characterized by abnormal accumulation of ceroid and lipofuscin in neurons and other cell types. Otto Christian Stengel first described neuronal ceroid lipofuscinosis in the medical literature as a juvenile-onset disorder with blindness and progressive dementia.1 A similar clinical entity was later described by Frederick Batten in 1903, and Walter Spielmeyer and Oskar Vogt in 1905.2-4
Prior to the discovery of causative genes associated with the disorder, the neuronal ceroid lipofuscinoses were classified based on age at onset (infantile, late infantile, juvenile and adult). More recently, their classification has been based on the mutated genes.5 The genetic basis for the classic juvenile-onset form is due to mutation in the CLN3 gene, located on chromosome 16.6 The gene codes for a lysosomal transmembrane protein of unknown function. In 81 to 85 percent of CLN3 disease, patients are homozygous for the 1.02kb deletion seen in our patient.6,7 Point mutations and insertions have also been found to be causative.7
JNCL has an estimated incidence of one in 25,000 births.8 The incidence is higher in northern European populations.8 The average age of onset is 4 to 7 years old. CLN3 disease is characterized by rapid visual decline secondary to retinal degeneration associated with progressive cognitive decline, seizures, motor dysfunction and, ultimately, death.9
Patients with CLN3 have intracellular deposition of lipopigment, resulting in loss of cells in the RPE, photoreceptor, outer nuclear and outer plexiform layers. Previous electron microscopy studies have shown that ocular damage starts with the photoreceptors and outer retina.10 Degeneration tends to progress from the macula to the periphery.10 On clinical examination, there are variable fundus findings associated with degeneration of the retina and optic nerve. Macular abnormalities include loss of foveal reflex, “bull’s-eye” maculopathy and macular orange pigment deposition.11,12 Mid-peripheral retinal pigmentary changes range from “bone spicule” pigmentary clumping to mild focal deposits.13 Secondary optic nerve atrophy and vascular attenuation are also common findings.13 A recent study found that patients diagnosed with JNCL may have macular striations.13 These striations may represent pathological degeneration or secondary inflammatory process within the internal limiting membrane and nerve fiber layer,13 and can be seen in many juvenile retinal dystrophies.
The initial diagnosis may be challenging due to non-specific retinal findings. Patients may be misdiagnosed as having Stargardt’s disease, retinitis pigmentosa or rod-cone dystrophy.11,12 Perhaps the most important clues to the diagnosis are rapid visual deterioration in a child accompanied by neurologic signs. Of note, the retinal dystrophy may be present before the onset of neurologic decline. In patients suspected of having a neuronal lipofuscinosis, genetic testing can be performed. Testing the five common mutations in CLN1, CLN2 and CLN3 may identify more than 70 percent of pediatric cases.14
There is currently no cure for the neuronal lipofuscinosis, although multiple treatments are being investigated.15,16 Previous studies have found autoantibodies present in cln3 -/- mice, suggesting an autoimmune component to the pathogenesis of the disease.17 There is a Phase II clinical trial under way evaluating the efficacy of mycophenolate mofetil in decreasing circulating autoantibodies. At the present time, management is mainly centered on potential enrollment in clinical trials as well as support and counseling for the patient and family.
JNCL is an autosomal recessive neurodegenerative disease presenting in childhood, characterized by relatively rapid vision loss and neurologic decline. The disease should be considered in any child less than 10 years old presenting with relatively rapid vision loss and signs of a retinal dystrophy. REVIEW
1. Stengel OC. Beretning om et maerkeligt Sygdomstilfaelde hos fire Sødskende I Nærheden af Röraas. Eyr 1826;1:347-52.
2. Batten FE. Cerebral degeneration with symmetrical changes in the maculae in two members of a family. Trans Ophthalmol Soc UK 1903;23:386-90.
3. Spielmeyer W. Über familiäre amaurotische Idiotien. Neurol Cbl 1905;24:612-20.
4. Vogt H. Über familiäre amaurotische Idiotie und verwandte Krankheitsbilder. Mschr Psychiatr Neurol 1905;18:161-71.
5. Williams RE, Goebel HH, Mole SE, et al. NCL nomenclature and classification. In: Mole SE, Williams RE, Goebel HH, eds. The Neuronal Ceroid Lipofuscinoses (Batten Disease). Oxford, Oxford University Press, 2011:20-3.
6. The International Batten Disease Consortium. Isolation of a novel gene underlying Batten disease, CLN3. Cell 1995;82(6):949-57.
7. Munroe PB, Mitchison HM, O’Rawe AM, et al. Spectrum of mutations in the Batten disease gene, CLN3. Am J Hum Genet 1997;61(2):310-6.
8. Zeman W. Studies in the neuronal ceroid lipofuscinosis. J Neuropathol Exp Neurol 1974; 33(1):1-12.
9. Collins J, Holder GE, Herbert H, et al. Batten disease: Features to facilitate early diagnosis. Br J Ophthalmol 2006;90(9):1119-24.
10. Traboulsi EI, Green WR, Luckenbach MW, de la Cruz ZC. Neuronal ceroid lipofuscinosis. Ocular histopathologic and electron microscopic studies in the late infantile, juvenile, and adult forms. Graefe’s Arch Clin Exp Ophthalmol 1987;225(6):391-402.
11. Bohra LI, Weizer JS, Lee AG, Lewis RA. Vision loss as the presenting sign in juvenile neuronal ceroid lipofuscinosis. J Neuroophthalmol 2000;20(2):111-5.
12. Horiguchi M, Miyake Y. Batten disease; deteriorating course of ocular findings. Jpn J Ophthalmol 1992;36(1):91-6.
13. Dulz S, Wagenfeld L, Nickel M, et al. Novel morphological macular findings in juvenile CLN3 disease. Br J Ophthalmol 2015. Epub ahead of print.
14. Zhong N. Neuronal ceroid lipofuscinoses and possible pathogenic mechanism. Mol Genet Metab 2000;71(1);195-206.
15. Kinarivala N, Trippier PC. Progress in the development of small molecule therapeutics for the treatment of neuronal ceroid lipofuscinoses (NCLs). J Med Chem 2015. Epub ahead of print.
16. Drack AV, Mullins RF, Pfeifer WL, et al. Immunosuppressive treatment for retinal degeneration in juvenile neuronal ceroid lipofuscinosis (juvenile Batten disease). Ophthalmic Genet 2015;36(4):359-64.
17. Seehafer SS, Ramirez-Montealegre D, Wong AM, et al. Immunosuppression alters disease severity in juvenile Batten disease mice. J Neuroimmunol 2011;230(1-2):169-72.


