Over a series of articles, I'm reviewing the causes, diagnosis and treatment options for patients with vertical diplopia. The initial discussion focused on contribution of the orbital muscles and clinical tests to assess this component of the diagnosis. (See the November 2007 issue, or visit the archives at revophth.com) This second installment will explore the anatomical pathways and activity and lesions that can cause vertical diplopia. Because of the complexity of this review, it will appear in two parts, with the second in next month's issue.
Identifying the Lesion
After correctly identifying the underacting eye muscle(s) in a person with vertical diplopia, you must decide if the culpable lesion is supranuclear, infranuclear or restrictive in origin. The more familiar infranuclear pathway simply consists of the lower motor neuron unit of cranial nerves 3, 4 and 6. It begins in the nucleus of each cranial nerve where a pool of motor neurons sprout axons that coalesce to form a cranial nerve that courses along the skull base to the orbit where they innervate their respective eye muscles. The supranuclear pathway, far less familiar to the practicing ophthalmologist, consists of the complex of neurons, axons and axonal interconnections that directly influence the lower motor neuron pools of the 3rd, 4th and 6th cranial nerves. Figure 1 depicts some of these neural connections in the cerebral hemispheres, deep cerebral structures, vestibular apparatus, cerebellum and the brainstem that eventually converge on the horizontal and vertical "gaze centers." Gaze centers generate the immediate prenuclear signal to the ocular motor neurons that execute conjugate horizontal and vertical gaze. The terms conjugate and gaze apply to the movement of both eyes together. The gaze centers that control conjugate horizontal and vertical gaze are located in different regions of the brainstem. The horizontal gaze center primarily resides in the caudal pons within the confines of the 6th nerve nucleus and an adjacent area called the paramedian, pontine reticular formation (PPRF). Neuronal activity in and destruction of the caudal pons in and around the PPRF and 6th nerve nucleus primarily affect conjugate eye movement along the horizontal plane.
The immediate supranuclear generators for vertical eye movement are located in the midbrain and diencephalon; specifically, dorsal, medial and anterior to the 3rd nerve nucleus in the vicinity of the mesencephalic pre-tectum and the rostral mesencephalic reticular formation (MRF). Single unit recordings have identified neurons within the MRF that discharge preferentially and are time-locked to fast eye movements with a vertical component.1 Approximately equal numbers of neurons in the MRF are activated by upward and downward gaze, but none are activated by horizontal eye movements.
Anatomically separate vertical gaze centers generate conjugate downward and upward eye movement. The rostral interstitial nucleus of the median longitudinal fasciculus (rostral i-MLF) appears to act as the immediate supranuclear generator for downward gaze. Studies have documented bilateral projections from the rostral i-MLF to the oculomotor complex, and paralysis of downward gaze with intact upward gaze has been reported with clinically observed and experimentally induced lesions in this region.2
Supranuclear paralysis of up gaze most commonly results from bilateral lesions of the pre-tectal region that involve fibers and nuclei of the posterior commissure. The same abnormality of eye movement may, on rare occasions, result from a single lesion of the posterior commissure, an observation that has been attributed to the profuse decussation of fibers within the commissure.3 Parinaud's syndrome or the syndrome of the pretectal plate is the classic example of an anatomical lesion that clinically produces a selective supranuclear palsy of conjugate up gaze with normal down gaze. Lesions in this area may also produce bilateral lid retraction, bilateral, pupillary light-near dissociation, retraction nystagmus and a skew deviation.
Lesions of the Supranuclear Pathway
The aggregates of prenuclear neurons within each "gaze center" receive two major forms of supranuclear input: 1) consciously driven signals that originate in the cerebral hemispheres, which initiate the volitional or conscious decision to move the eyes horizontally or vertically; and 2) reflex-driven signals from the vestibular apparatus that authorize involuntary eye movement in response to changes in head posture. For instance, the conscious decision to move the eyes in the vertical plane begins in the cerebral cortex. From here bilateral projections to the MRF reach the rostral i-MLF for conjugate down gaze or to the pretectal plate for conjugate up gaze. These neuronal projections are modified along the way from interconnections between the superior colliculus, thalamus and cerebellum.
Reflex-driven eye movements are initiated and generated in the peripheral vestibular organs. Signals from the semicircular canals and utricles travel centripetally along the vestibular nerve where they are modified by complex interconnections between the cerebellum and vestibular nucleus. Eventually they terminate on the lower motor neurons of the 3rd, 4th and 6th cranial nerves. The vestibulo-ocular reflex, for example, is a three-neuron arc that begins with an afferent sensory end organ, the semicircular canals and utricle. Detecting motion, the semicircular canals and utricle generate a neural signal along the vestibular nerve which synapses with the motor neurons of the vestibular nuclear complex in the caudal brainstem and relays the neural signal to the contralateral lower motor neuron pools of cranial nerves 3, 4 and 6. From here efferent motor fibers of each cranial nerve project to and synapse with the appropriate extraocular muscles. The vestibulo-ocular reflex serves vision by generating conjugate smooth eye movements in response to head movements, specifically, a conjugate eye movement that moves at the precise velocity, but in the opposite direction to head movement, thus allowing the eyes to remain stationary in space during head movement. Image stability on the retina is maintained despite horizontal, vertical or torsional translations of the head.
Lesions of the supranuclear pathway generally affect the ocular movement of both eyes in a symmetrical manner. In most supranuclear palsies of eye movement, a conscious person may not be able to volitionally move his eyes conjugately in a particular direction, but the limitation of conjugate eye movement can be overcome by the doll's eye maneuver or the Bell's reflex, if the vestibular-ocular connections are intact. The doll's eye maneuver is accomplished by having the patient maintain fixation straight ahead while either the patient or the examiner turns, flexes or hyperextends the patient's chin. Flexing the chin evokes conjugate up gaze by reflex, and depressing the chin evokes conjugate down gaze via the vestibular-ocular reflex. The
Because most supranuclear lesions of eye movement affect the motility of both eyes equally, the eyes remain parallel, and double vision is not a prominent complaint. Three important exceptions to this rule deserve description: internuclear ophthalmoplegia, skew deviation and an uncommon syndrome entitled supranuclear paralysis of monocular elevation.
Internuclear Ophthalmoplegia
Recall that both consciously driven and reflex-driven excitatory impulses pass from the PPRF to the oculomotor neurons, i.e., neurons within the 3rd, 4th and 6th nucleus, in contrast to the more specific oculomotor neurons that reside only in the 3rd nerve nucleus. For horizontal gaze, the PPRF "excites" the 6th nerve nucleus, which contains two separate populations of lower motor neurons. One set gives rise to the 6th cranial nerve and innervates the ipsilateral lateral rectus muscle; another set, the internuclear neurons of the 6th nerve nucleus, whose axons cross the midline at the level of the nucleus, ascend in the median longitudinal fasciculus (MLF), and synapse in the contralateral medial rectus subnucleus of the oculomotor complex. The axons from the medial rectus subnucleus of the 3rd nerve nuclear complex supply the excitatory impulses to the ipsilateral medial rectus muscle. 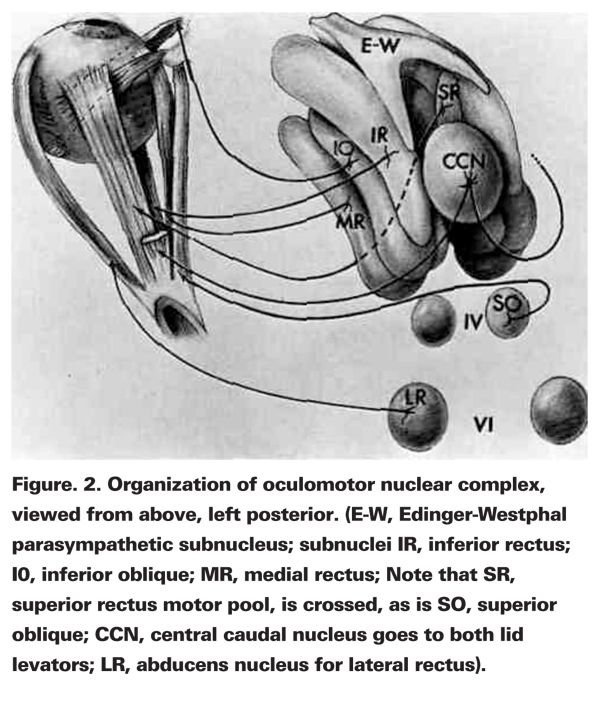
A lesion of the MLF is tantamount to a supranuclear lesion of the oculomotor nucleus, specifically the medial rectus subnucleus of the oculomotor nucleus. Damage to the MLF blocks impulses to the ipsilateral medial rectus. Consequently, the eye ipsilateral to the MLF lesion cannot adduct spontaneously, or on command, or in response to the doll's maneuver. But, because there is another supranuclear input to the medial rectus subnucleus from a convergence center, the affected eye will adduct during convergence. Hence, an INO sharply localizes to an ipsilateral lesion of the MLF. Common causes include multiple sclerosis, stroke, trauma and neoplasm. Although INO mostly causes horizontal diplopia, patients with an INO will often show a skew deviation.
Skew Deviation
Skew deviation refers to an acquired vertical dissociation of the eyes of supranuclear etiology. In addition to a vertical misalignment, a skew may also include a torsional and horizontal component. Subjective vertical diplopia is almost always present in the primary position, although the vertical misalignment may be comitant, incomitant, paroxysmal, alternating or periodic.4-10 Some of the incomitant skew deviations may be difficult to distinguish from a vertical extraocular muscle palsy, 4th nerve palsy or partial 3rd nerve palsy, especially when there is an accompanying element of ocular torsion and head tilt.11-13 In one report of 100 cases of skew deviation, many of the patients even met the three-step criteria for a superior oblique muscle palsy.6
When a skew deviation is incomitant, it can usually be differentiated from vertical eye muscle imbalance by the following signs:
1) Skew almost always occurs with other signs of brainstem and cerebellar dysfunction.
2) The amount of incomitance found with a skew is substantially less than that found with a vertical eye muscle palsy.
3) The amount and direction of the cyclotropia, when present, is small and it may be inconsistent with the vertical muscle palsy diagnosed by the three-step test.
4) Ocular rotations do not show as much overaction or underaction as seen in vertical muscle palsy.
5) Like other syndromes caused by a supranuclear lesion, the apparent limitation of vertical eye movement with skew can be overcome by the doll's eye maneuver or
Skew deviation has been observed with lesions of the vestibular organ, vestibular nerve, cerebellum, midbrain, pons or medulla, or as a manifestation of increased intracranial pressure.6,9,14-16 In the aforementioned report of 100 patients with skew, approximately 20 percent had midbrain or pretectal lesions, 60 percent had pontine damage and 20 percent demonstrated medullary signs. Patients with the more caudal brainstem lesions were more likely to show a minor vertical separation which varied with head position, but there were no consistent differential features of skew resulting from lesions of various levels of the brainstem. Patients with the syndrome of internuclear ophthalmoplegia were more likely to show an ipsilateral hypertropia, unless the lesion was medullary, in which case the ipsilateral eye was more commonly hypotropic.
Supranuclear Paralysis of Monocular Elevation
Two separate studies described a total of eight patients with an acquired supranuclear paralysis of monocular elevation.17,18 The outstanding characteristics of this syndrome included: sudden-onset elevation palsy in one eye; orthotropia in primary position with no subjective diplopia in primary position; a limitation of monocular elevation that was the same whether the eye started from primary position, adduction or abduction; and full ocular elevation with the Bell's or doll's eye maneuver. Occasionally, pupillary abnormalities, convergence weakness and paresis of depression of the affected eye were present, but no evidence of ptosis, lid retraction, inflammatory signs, exophthalmus, enophthalmus or a positive forced ductions or a positive Tensilon test. The findings were attributed to a lesion of the high midbrain interrupting the supranuclear inputs to the two eye muscles that elevate the globe, i.e., the superior rectus and inferior oblique. The contention of the authors of the first, that the site of the presumed lesion must be pre-tectal, received support from the latter, whose author observed similar clinical findings in a man with a metastatic bronchogenic carcinoma that involved a unilateral segment of the upper midbrain.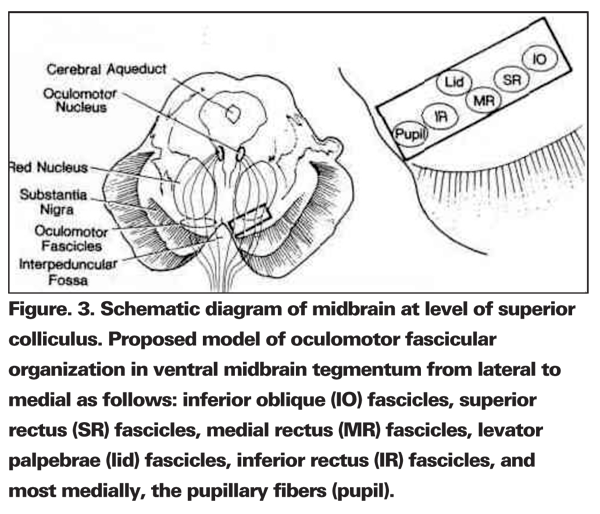
The relatively uncommon syndrome of supranuclear paralysis of monocular elevation bears a superficial resemblance to the more common double elevator palsy syndrome. In both, there is a conspicuous absence of subjective diplopia in primary position, and the monocular elevation weakness is the same starting from primary position, abduction or adduction. The double elevator syndrome, however, consists of three types: 1) the most common are those with prominent inferior rectus restriction who show positive forced ductions to elevation and normal saccades in the field of action of the superior rectus muscle; 2) elevator weakness, negative forced ductions and reduced saccadic velocities in upward movement of the affected globe; and 3) combinations of types 1 and 2. In a report of 15 patients with this syndrome, investigators found that 12 of them had positive forced ductions and normal upward saccadic velocity, strongly suggesting a restrictive ophthalmopathy and a "peripheral mechanism."
Distinguishing features of the double elevator syndrome are that it is usually congenital and often accompanied by ptosis ipsilateral to the apparent upgaze palsy. In fact, the patient often assumes a chin-up position to compensate for the ptosis, since a primary position hypotropia is relatively uncommon.
Anatomy and Lesions of the Infranuclear Pathway
1. Oculomotor Nerve. The 3rd cranial nerve supplies the levator palpebrae superiorus, superior rectus, medial rectus, inferior rectus and the inferior oblique muscles, and contains visceral motor nerves that innervate the parasympathetic supply to the pupilloconstrictor and ciliary muscles. A complete, unilateral 3rd nerve palsy is characterized by complete ptosis, a large unreactive pupil, and paralysis of ocular elevation, adduction and depression. Partial 3rd nerve palsies show less extensive involvement.
The nuclear complex of the oculomotor nerve, lying beneath the aqueductal gray matter of the rostral midbrain at the level of the superior colliculus, consists of two midline, unpaired visceral nuclei and four paired nuclei. The anterior midline nucleus, the Edinger-Westphal nucleus, contains the motor neurons that supply the pupil; the more posterior central caudal nucleus contains the motor neurons that supply both levator palebrae superiorus muscles. Axons from the superior rectus subnucleus decussate at the nuclear level to supply the contralateral superior rectus muscle. All the other oculomotor subnuclei supply ipsilateral extraocular muscles: the dorsal subnucleus supplies the inferior rectus; the intermediate nucleus supplies the inferior oblique; and the medial rectus muscle is supplied by axons that arise from at least three separate motorneuron pools within the oculomotor complex.
This discrete layering of motorneuronal pools within the oculomotor nuclear complex can be used to explain the clinical manifestations of nuclear lesions. For example, a nuclear lesion has been shown to cause isolated bilateral internal ophthalmoplegia (pupillary areflexia), isolated bilateral ptosis or an isolated palsy of one inferior rectus muscle.21-23 More commonly, a nuclear lesion will produce a unilateral palsy of the 3rd nerve in conjunction with weakness of the ipsilateral and contralateral superior rectus muscles, with bilateral ptosis.
The axons from each oculomotor subnucleus form a loose bundle of nerve fascicles in the midbrain that course ventrally through the median longitudinal fasciculus, superior cerebellar peduncle, red nucleus and cerebral peduncle to emerge from the brainstem as a single nerve trunk in the interpeduncular cistern. Fascicular lesions of the 3rd nerve produce symptoms and signs that are strictly ipsilateral. Due to the relative anatomic separation of the 3rd nerve, fascicles within the midbrain fascicular lesions often cause partial rather than complete 3rd nerve palsies. Examples of fascicular lesions include: isolated inferior oblique paresis;24 a unilateral, fixed, dilated pupil unassociated with other neurologic signs;25 isolated paresis of the inferior oblique, superior rectus, medial rectus, and levator muscles, with sparing of the ipsilateral inferior rectus and pupil;26,27 and finally, paresis of the inferior oblique, superior rectus, medial rectus, levator and inferior rectus muscles, with pupillary sparing.26,28 From an analysis of these reports, one group attempted to establish a topographic organization of the individual oculomotor fascicles in the midbrain.24 From lateral to medial, these authors pictured the inferior oblique, superior rectus, medial rectus and levator palpebrae, inferior rectus, and most medial pupillary fibers. If in this organizational plan, the levator fibers were lateral to the medial rectus fibers, a local midbrain lesion could conceivably produce a pattern of abnormal ocular motility that mimics a "superior division" 3rd nerve palsy.29-31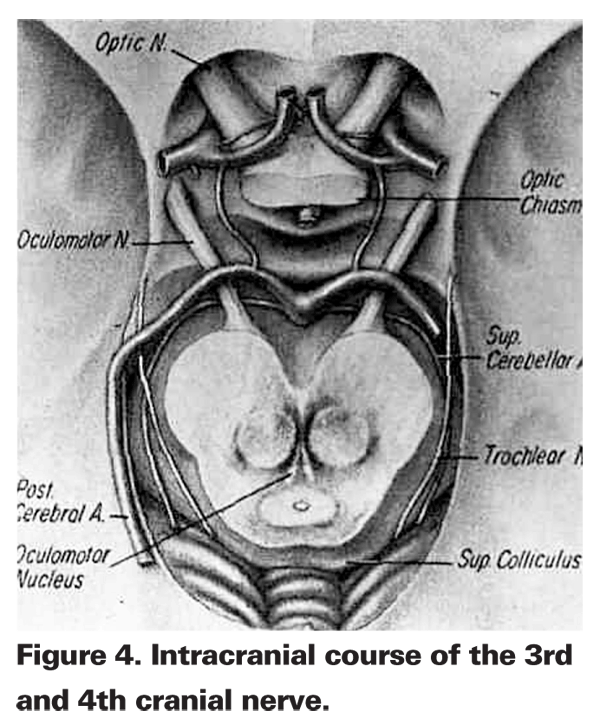
Fascicular lesions are best recognized "by the company they keep." Classic syndromes have been described. One author described three patients, each of whom had an oculomotor palsy and contralateral hemiparesis, "rubral" tremor and involuntary movements.32 An autopsy of one of these patients revealed a tuberculoma within the right cerebral peduncle in the mesencephalon, obliterating the 3rd nerve, ipsilateral red nucleus, substantia nigra and cerebral peduncle. A "rubral" or midbrain tremor contralateral to a 3rd nerve palsy is caused by a lesion of the 3rd nerve where it courses through the red nucleus. The tremor is usually unilateral and may be present at rest or with intention. It is classically described as a resting tremor that worsens on adopting a fixed posture or on attempted movement.
Claude's syndrome consists of a 3rd nerve palsy with contralateral ataxia and choreiform movements. In this syndrome, the substantia nigra and cerebral peduncle are spared, but it involves the subthalamic nucleus, crossing fibers of the superior cerebellar peduncle and the medial longitudinal fasciculus.33 Chorea, coined from the Greek word meaning dance, describes fidgeting movements of the hands and the unstable dance-like gait, best exemplified by advanced cases of the infectious and degenerative choreas. In choreiform tremor, variability is the rule, a particular movement being repeated only rarely. Most often, choreic movements are seen in the face and head and/or the distal parts of the upper extremities.
Another study described the features of tumorous lesions of the quadrigeminal plate in patients with increased intracranial pressure.34 And Weber's syndrome comprises an oculomotor palsy and contralateral hemiparesis.35 The culpable lesion in a Weber's syndrome encompasses the 3rd nerve and cerebral peduncle, through which the descending corticospinal tract passes. Therefore, the lesion may be intra- or extra-axial, as it may affect the 3rd nerve within the cerebral peduncle or just after it emerges from the cerebral peduncle into the interpeduncular cistern.
The subarachnoid portion of the 3rd nerve passes between the posterior cerebral and superior cerebellar arteries and then courses anteriorly to pierce the posterior wall of the cavernous sinus. The 3rd nerve runs along the lateral wall of the cavernous sinus just superior to the trochlear nerve, and then continues forward through the superior orbital fissure. In the cavernous sinus, the 3rd, 4th and 6th cranial nerves are extradural. Either in the anterior portion of the cavernous sinus or within the superior orbital fissure, the 3rd nerve bifurcates into a superior and inferior division. Branches from the superior division supply the superior rectus muscle and the levator palpebrae superiorus. The inferior division innervates the medial rectus, inferior rectus and inferior oblique muscles and also transmits parasympathetic axons, via the ciliary ganglion, to the pupilloconstrict or muscle of the iris and the ciliary body.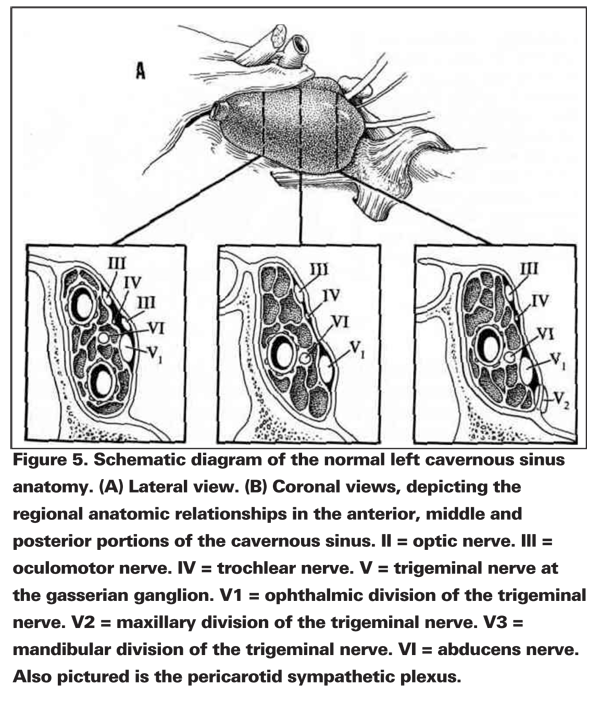
The most common lesions affecting the subarachnoid portion of the 3rd nerve are trauma, rupture or expansion of a posterior communicating-internal carotid artery aneurysm and small vessel disease, usually from diabetes mellitus or systemic hypertension. Trauma and aneurysms generally tend to produce a complete 3rd nerve palsy, although it should be emphasized that "pupil-sparing" 3rd nerve palsies have been described with aneurysmal compression. The classic dilemma—when to aggressively study and when to defer studies in a person with a new-onset 3rd nerve palsy—formed the basis of a thoughtful report.36 These authors divided 41 patients with oculomotor palsies into several groups: 28 with a presumed microvascular infarction of the third nerve; 10 with an angiographically proven aneurysm (seven posterior communicating and three intracavernous carotid aneurysm); two with intracavernous meningiomas, and one with CNS lymphoma. By culling the results of previous studies and their observations, the group reached the following conclusions:
• Intradural cerebral aneurysms, which most commonly arise at the junction of the posterior communicating and internal carotid arteries, tend to produce pupillary involvement at presentation. Hence, a partial or complete 3rd nerve palsy that involves the pupil, i.e., a large and unreactive pupil, should be evaluated emergently. In patients older than 20 years of age, this would include neuroimaging studies like MR angiography, CT angiography and traditional catheter angiography. The presence or absence of headache, neck stiffness, confusion and other signs of subarachnoid hemorrhage should not alter these management decisions.
• Patients with complete ptosis, and paralysis of ocular elevation, adduction and depression, in whom the pupil shows normal or near-normal size and reactivity at presentation, should be managed more deliberately, since they almost always have had a microvascular infarction of the 3rd nerve. This rule is particularly true if the patient is older than 50 years of age and has no symptoms of subarachnoid hemorrhage.
• A purely superior-division 3rd-nerve palsy, which will spare the pupil, is rarely caused by microvascular infarction of the third nerve and mandates a careful search for a compressive lesion.36,37
• Sixty-eight percent of the patients with "vascular" 3rd-nerve palsies improved by one month, 96 percent by two months and 100 percent by three months.
In another analysis of patients with pupil-sparing 3rd-nerve palsies with internal carotid-posterior communicating artery aneurysms, it was observed that the pupil may be spared initially with aneurysmal compression, but invariably becomes involved over one week of observation.38 Thus, if a patient presents with signs of a pupil-sparing 3rd-nerve palsy a week or more after the onset of symptoms, it is highly doubtful that he has an intracranial aneurysm.
Microvascular disease of the 3rd nerve produces a syndrome of pupillary sparing. A true pupil-sparing 3rd-nerve palsy should be confined to paralysis (complete, not partial weakness) of ocular adduction, elevation and depression, with normal or near-normal pupillary function. Several neuroanatomic39 and neuropathologic studies40-42 of this syndrome have shown small watershed microinfarctions of the 3rd-nerve trunk in either the subarachnoid or cavernous segments. Microscopically, the infarction seems to preferentially affect the deeper, central portions of the 3rd nerve, sparing the superficially positioned pupillary fibers.39 Prior to the advent of CT and MRI scanning, all sudden-onset pupil-sparing 3rd-nerve palsies were attributed to a lesion of the extraparenchymal portion of the oculomotor nerve.40-42 Subsequent MRI and CT data have clearly documented small intraparenchymal lesions of the midbrain in such patients,28,43 prompting several reviewers to recommend a noninvasive imaging study in all cases of 3rd-nerve dysfunction. 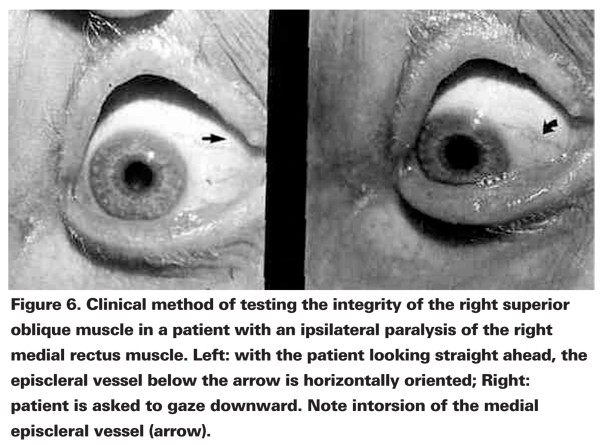
Aberrant regeneration of the oculomotor nerve most commonly occurs after severe head trauma or as a result of rupture or sudden expansion of an internal carotid-posterior communicating artery aneurysm. Both of these conditions typically disrupt the subarachnoid portion of the 3rd nerve proximal to its bifurcation. Aberrant regeneration results in synkinetic movements of muscles innervated by both the superior and inferior divisions. In its classic form, aberrant regeneration of the oculomotor nerve is characterized by elevation of the involved lid on adduction, lowering of the lid during abduction, elevation of the lid on depression of the eye, absence or only partial vertical eye movement (often with adduction on the attempt), poor pupillary response to a light stimulus that is often segmental, and constriction of various pupillary sphincter segments on adduction, depression or elevation of the globe. On rare occasions, when there is a divisional palsy of the 3rd nerve, aberrant regeneration may be limited to the structures innervated by that division. I reported such a patient with aberrant regeneration limited to the extraocular muscles innervated by the inferior division of the 3rd nerve. Spontaneous aberrant regeneration of the 3rd nerve, i.e., signs of aberrant regeneration without a history of head trauma or aneurysm, mandates a search for a slow-growing lesion like an intracavernous meningioma.
Intracavernous 3rd-nerve involvement is frequently accompanied by involvement of the closely adjacent sympathetic, abducens, trochlear and trigeminal nerves. Altered facial sensation in the form of paresthesias, hypalgesia, formications and pain may be the salient symptoms of trigeminal nerve involvement. If present in the distribution of the ophthalmic division of the trigeminal nerve (V-l), along with an ipsilateral partial or complete 3rd-nerve paresis, the lesion tends to involve the anterior cavernous sinus or superior orbital fissure; sensory symptoms in the maxillary division (V-2) of the trigeminal nerve, with 3rd-nerve palsy, points to a posterior lesion of the cavernous sinus.
Either just before or just after the 3rd nerve enters the superior orbital fissures, it splits into a superior and inferior division. A superior division palsy is characterized by ptosis and weakness of ocular elevation, due to involvement of the superior division's branches to the superior rectus and levator palpebrae superiorus.
An inferior division palsy would manifest as pupillary paralysis with weakness of the inferior and medial rectus and inferior oblique with pupillary paralysis; the lid and ability to elevate the globe would be unaffected. Lesions of the orbital apex and orbital segments of the 3rd nerve tend to produce the foregoing signs with proptosis and loss of vision from optic nerve involvement.
2. Trochlear nerve. The trochlear nerve only innervates the superior oblique muscle. The nucleus of the trochlear nerve, located in the tegmentum of the midbrain at the level of the inferior colliculus, is located near the midline just ventral to the cerebral aqueduct. Axons from the nucleus course dorsally around the cerebral aqueduct and decussate within the superior medullary velum where they exit the midbrain dorsally and continue curving forward and around the dorsolateral midbrain to emerge between the posterior cerebral and superior cerebellar arteries. Running anteriorly, the 4th nerve pierces the dura at the angle between the free and attached borders of the tentorium cerebelli and eventually enters the cavernous sinus. After traversing the superior orbital fissure, the nerve crosses medially close to the roof of the orbit, and runs diagonally across the levator palpebrae and superior rectus muscle to reach the superior oblique muscle. Here the nerve divides into three or more branches and enters the superior oblique muscle along the proximal one-third.
The signs of a unilateral lesion of the 4th nerve include a positive three-step test and excyclodeviation of the affected eye. With lesions that are proximal to the 4th nerve's decussation, the contralateral superior oblique muscle is weak; lesions distal to the 4th nerve's decussation produce an ipsilateral palsy of the superior oblique. Proximal lesions may be intra- or extraparenchymal. With an intra-parenchymal lesion there may be other findings, such as: thermoanalgesia (relative loss of pain and temperature sensation) of the face, arm, trunk and leg; limb ataxia; Homer's syndrome; and, on rare occasions, an afferent pupillary defect. Distal lesions are far more common. Head trauma, small vessel disease and space-occupying lesions comprise the most common etiologies.
Certain nuances of superior oblique muscle palsy deserve emphasis. If the primary action of the superior oblique is to depress the eye in adduction, what happens if the globe cannot be adducted? How then do you test the integrity of the ipsilateral 4th nerve in the presence of medial rectus paralysis, e.g., from a complete 3rd-nerve palsy? The question is more than rhetorical, since the neuroanatomic localization and management of a patient with an isolated 3rd-nerve palsy differ from that of the patient with a combined 3rd- and 4th-nerve palsy.
In primary position the action of the superior oblique muscle is to intort the globe. If adduction is paralyzed, you have only to observe one of the episcleral vessels as the patient attempts to look down. If the vessel intorts, the superior oblique muscle (and the trochlear nerve) must be intact. Failure to note intorsion may indicate involvement of at least two eye muscles or two cranial nerves.
The term "masked 4th-nerve palsies" applies to the situation of bilateral, but asymmetric superior oblique muscle palsies. If the asymmetry is significant the milder paresis may not be recognized with routine testing. Particular attention should be paid to the patient with a post-traumatic 4th-nerve palsy, because bilateral involvement should be assumed until proved otherwise. In patients with traumatic 4th-nerve palsies, check for a reversal of the hypertropia in the oblique fields of gaze—gaze down and up to the right and left—and quantify the excyclodeviation with the double Maddox rod test. For example, a 24-year-old woman complained of diplopia after closed head trauma. Subjectively, images were separated obliquely from each other, especially in right head tilt. Results of a prism cover test are depicted in Figure 7. Based on the three-step test alone, there was evidence of a right superior oblique paresis. But further testing showed that in the oblique fields of gaze a small left hypertropia was also observed, and the double Maddox rod test revealed 13 degrees of excycloduction. The correct diagnosis is a bilateral superior oblique palsy. Although measuring strabismus in the oblique fields of gaze can be technically difficult, at least do a qualitative cover test in the oblique fields of gaze to help rule out a bilateral and "masked" superior oblique palsy. The double Maddox rod test provides additional evidence of bilateral superior oblique muscle involvement. With unilateral superior oblique muscle palsy the amount of excyclotropia measured with the double-Maddox rod rarely exceeds 8 degrees; with bilateral involvement it is often greater than 12 degrees.
Neuromuscular Junction Defects
The neuromuscular junction is composed of the following anatomic units: the presynaptic membrane at the distal nerve; a synaptic cleft or space between distal nerve and muscle; and a receptor-laden postsynaptic membrane that anatomically belongs to the extraocular muscle. The biotransmitter acetylcholine (AcH) is synthesized, packaged and released from the presynaptic membrane into the synaptic cleft in the form of packets or quanta. During periods of muscular relaxation, small numbers of quanta are released at a relatively slow rate, about one per second. Each individual quantum of AcH binds to a receptor on the postsynaptic membrane, generating a tiny electrical potential called a miniature end plate potential (MEPP). When a greater innervational signal is required; e.g., to generate a muscular contraction, more quanta are released. The amplitude of the MEPP summates, resulting in a larger electrical charge at the postsynaptic membrane called an end plate potential (EPP). If the EPP exceeds the excitability threshold of the muscle's postsynaptic membrane, a muscle action potential produces a muscular contraction. The muscle action potential is an all-or-none response, while the MEPPs and EPPs are graded responses.
The efficiency of neuromuscular transmission depends on many factors, including the bioavailability of AcH; that in turn is directly related to the nerve's ability to synthesize, package and release the necessary quanta of AcH and on the binding efficiency of the postsynaptic membrane. Adequate concentrations of circulating calcium catalyze the binding. Consequently abnormally low or high levels of calcium can interfere with normal neuromuscular transmission by affecting the size of the EPP.
Certain anesthetics, cardiac medications, antibiotics and other drugs should be used cautiously in patients with disorders of the neuromuscular junction, in particular myasthenia gravis, because they may significantly impair neuromuscular transmission. These include succinylcholine, curare vecuronium, quinine, quinidine, procaineamide, propranolol, calcium channel blockers, polymixins, tetracyclines, penicillamine, aminoglycosides, ciprofloxacin and telithromycincan. While the synthesis, packaging and release of AcH appears to be intact in myasthenia gravis, the bioavailabilily of AcH varies because of the presence of a blocking antibody that alters the architecture of the postsynaptic membrane and the efficiency of neuromuscular transmission.
The accurate diagnosis of myasthenia gravis is dependent on two factors: the examiner's index of suspicion and a knowledge of the technique and pitfalls of the intravenous Tensilon test. The following clinical situations warrant doing a Tensilon test: a patient with ptosis and/ or ophthalmoplegia whose symptoms and signs fluctuate, particularly if he improves with sleep or rest and worsens with fatigue, exposure to bright light or during the eye examination; a case in which ptosis fluctuates, or alternates sides; a case of ptosis and/or diplopia in association with constant or varying strength of speech, swallowing or the extremities; a case of ptosis or ophthalmoplegia associated with symptomatic or asymptomatic weakness of the orbicularis oculi muscle(s); and when ptosis or ophthalmoplegia occur without an obvious explanation during pregnancy, the postpartum, following other surgical procedures or major illnesses, and in persons with thyroid ophthalmopathy or collagen vascular disease.
Prior to doing an intravenous Tensilon test, be sure to inquire if the patient has cardiac disease, particularly a cardiac arrhythmia, because intravenous Tensilon may induce a profound bradycardia, with consequent syncope, nausea and vomiting. In persons with cardiac disease or in patients over age of 55, it is best to perform the intravenous Tensilon test in a hospital or emergency room setting. In all age groups, it should be done with a vial of atropine nearby to avert the symptoms of severe and symptomatic bradycardia.
The intravenous dose of Tensilon should be 0.15 mg/kg in children and up to 10 mg total in adults. A small test dose of Tensilon should be given first and the response assessed 30 to 45 seconds later. If there are no untoward side effects, incremental injections of 2 to 3 mg can be given until you note either improved strength in the patient, or symptoms or signs of cholinergic toxicity, such as sweating, nausea, faintness or abdominal cramping.
A pitfall of the intravenous Tensilon test: The therapeutic effect of Tensilon on ocular motility may be obscured for many reasons. Variables include: the patient's subjective interpretation of his pharmacologic response to Tensilon and the examiner's biased or unbiased interpretation; the drug has a transient active duration; the so-called "perverse" Tensilon response, in which a paradoxical worsening of some aspects of motility is associated with improvement in other aspects; the temporal variability of the disease; and some people with ocular or generalized myasthenia gravis may not respond to the drug.
To avoid these pitfalls, it is extremely important to have an objective sign of lid or eye muscle weakness prior to the drug's administration. Measuring the palpebral aperture and quantifying the results of a prism-cover test serve this purpose. Comparing these numeric data before and after the drug's administration helps to avoid examiner and patient bias.
Restrictive Factors
The clinical clues of a restrictive ophthalmopathy are listed in Table 1. According to Sherrington's law of reciprocal innervation, ductional eye movement or the movement of one eye is accomplished by simultaneous and equally graded contraction of the agonist and relaxation of the antagonist eye muscles. For instance, movement of the right eye to the right is accomplished by an excitatory innervational signal to the right lateral rectus and a concomitant inhibitory signal to its direct antagonist, the right medial rectus. The contraction of one muscle plus the simultaneous relaxation of its antagonist allows the eye to move quickly, smoothly and accurately towards fixation. Sherrington's law applies only to the muscles of one eye, the agonist and antagonist.
It follows that limitation of eye movement can be caused by denervation of the agonist or lack of inhibition of the antagonist eye muscle. The latter is referred to as a restrictive ophthalmopathy, a conclusion that can usually be reached by the forced duction test. The performance and interpretation of forced ductions can be obscured if the patient is too young, inattentive or squeamish to have his eyes manipulated, or by the presence of a very small limitation of eye movement. In these settings, other clinical techniques need to be applied.
Some investigators suggest measuring the intraocular pressures in primary gaze and in the direction of presumed weakness.44 In the presence of restriction, the healthy agonist muscle allegedly contracts against the pull of a tight antagonist, and intraocular pressure rises. This increased IOP in the direction of presumed gaze limitation compared to the IOP in primary position is considered by some to be a reliable sign of a restrictive ophthalmopathy.
Qualitatively and quantitatively judging the speed of saccadic eye movement can also be a useful and sensitive method of differentiating between denervational versus restrictive limitations of eye movement. Saccadic velocities are reportedly normal or near normal in restrictive ophthalmopareses,31,45-47 while they are abnormally slow in acute lesions of the 3rd, 4th and 6th cranial nerves. Patients with myasthenia gravis, interestingly, have been found to have very fast saccadic eye movements.
Refixation between two points in space is accomplished by a quick, ballistic, fast eye movement called a saccade. Saccades may be horizontal, vertical or oblique. The recti muscles—both the horizontal and vertical recti—appear to be primarily responsible for saccadic eye movements. With rectus muscle weakness from denervation, the resulting saccade is proportionately slow. However, with restrictive ophthalmopathy, saccades remain rapid, as has been shown in thyroid ophthalmopathy,46 orbital floor fracture,48 Brown's syndrome47 and rectus muscle contracture following paresis of the antagonist.19
Judging saccadic velocities, measuring the intraocular pressure in different directions of limited eye movement and performing the forced ductions test are the primary clinical tools we have to differentiate a restrictive ophthalmopathy.
Orbital Blow-out Fractures
The mechanism of orbital blow-out fractures is thought to be a sudden expansion of the intraorbital pressure being released centrifugally by fracture of the surrounding thin orbital bones. The orbital floor is thinnest posteriorly, where the thin bone immediately in front of the inferior orbital fissure is further weakened by the inferior orbital groove and canal. The orbital floor just medial to the infraorbital groove and immediately in front of the inferior orbital fissure is understandably the site of least resistance.
Orbital floor fractures in this location may incarcerate the inferior rectus muscle and its surrounding tissue. Larger defects may entrap the inferior oblique muscle or the relatively exposed nerve to the inferior oblique which enters the middle portion of the inferior oblique muscle after following the lateral border of the inferior rectus muscle. The ocular motility disorder of an orbital floor fracture can be caused by contusion injury of the orbital nerves or muscles, or incarceration, laceration, avulsion and hemorrhage into these structures.
Although protean patterns of ocular motility may result from orbital floor fractures, the most common motility disturbance is a monocular restriction of up gaze with positive forced ductions. In the acute phase, orbital floor fractures may simulate an oculomotor nerve palsy, since the affected eye may show vertical gaze limitation (from injury to both the inferior rectus and inferior oblique), pupillary paralysis (from denervation or direct ocular contusion) and "ptosis" as the patient squints to avoid double vision. The diagnosis can usually be clarified by the forced duction test.
Fracturing the central portion of the orbital floor near and within the inferior orbital groove frequently produces numbness in the distribution of the infraorbital nerve because of associated fractures of the zygomaticomaxillary suture and infraorbital foramen. Prolapse of the inferior orbital contents into the maxillary sinus also leads to enophthalmos and pseudoptosis, and in these cases, appropriate films show fluid levels in the maxillary sinus.
Dysthyroid Ophthalmopathy
Dysthyroid ophthalmopathy may occur in four types of thyroid disease: Graves' disease (thyrotoxicosis, goiter and exophthalmos); euthyroid Graves' disease; autoimmune thyroiditis; and hypothyroidism. Ocular symptoms may precede, occur with or follow by many years the systemic manifestations of hyper- or hypothyroidism, or they may occur in the absence of any other symptom of a dysthyroid state. The eye symptoms may be fulminant or subtle, and at times mimic a paralytic strabismus, especially when small amounts of strabismus make the interpretation of forced ductions test difficult to assess. Horizontal or vertical diplopia can be the earliest and only manifestation of dysthyroid ophthalmopathy for many months. In one subgroup of thyroid patients, in a condition referred to as euthyroid ophthalmopathy, there are clinical symptoms and signs of endocrine ophthalmopathy, but no confirmatory serological abnormalites of glandular dysfunction.
The eye muscles of patients with dysthyroid ophthalmopathy are enlarged, firm and rubbery, due to interstitial edema, endomysial fibrosis and a heavy infiltrate of collagen, mucopolysaccarides, lymphocytes and plasma cells. These changes both physically enlarge the eye muscles and affect the eye's position and motility. Such histopathologic changes seem to preferentially affect the inferior, medial and superior rectus muscles in that order of frequency; hence, the commonest patterns of abnormal ocular motility include unilateral or bilateral limitation of up gaze, lateral gaze, or down gaze, respectively.
If you suspect the diagnosis of thyroid ophthalmopathy, you can turn to a non-contrast CT or MRI scan, or standardized ophthalmic echography (SOE) for confirmation. CT and MRI should incorporate axial and coronal views of the orbit to clearly show the horizontal and vertical recti muscles. Better views of the medial and lateral recti are seen on the axial sections, while the superior and inferior recti are more clearly depicted on the coronal and sagittal views. Both CT and MRI depict the extraocular muscles in at least two planes, and the examiner must decide if the eye muscles are enlarged. Granted, there are established criteria for normal muscle size,49 but the final determination is still subjective. In contrast, a technically proficient ultrasonographer can obtain reproducible, quantitative and frequently histopathologically specific data regarding the size and composition of the extraocular muscle. SOE can also be used to identify incipient compression of the apical optic nerve in this patient population. Not all physicians have access to a technically proficient ultrasonographer. I have spent a lot of time improving my echography skills, and with all due modesty have reached the conclusion that SOE may be the only examination necessary to confirm many orbitopathies, including subtle thyroid eye disease.50 Certainly, the virtues of its low cost, convenience and low risk to the patient are obvious. I find it to be an extremely valuable tool in the differential diagnosis of a restrictive ophthalmopathy.
Brown's Syndrome
When elevation in adduction is limited, but upward eye movement from the primary position and from abduction are full, Brown's superior oblique tendon syndrome is a likely cause. The first form of Brown's syndrome was originally described as a congenital and permanent deficit, and it was related to a short anterior tendon sheath of the superior oblique tendon. Years later, Harold Whaley Brown modified his original thesis and allowed for other possible forms; namely intermittent; acquired and those which recover spontaneously with time.
Despite attempts to nosologically subclassify various subtypes of Brown's syndrome, the clinical features have remained relatively constant. It is almost always a unilateral deficiency of elevation in the adducted position. The inability to elevate the globe diminishes in primary upgaze and nearly or completely disappears in abduction. This change in ocular elevation between adduction and abduction distinguishes Brown's syndrome from double elevator syndrome, supranuclear palsy of monocular elevation and congenital fibrosis; in blow-out fracture a marked limitation of elevation is present in all upgazes. The severity of Brown's syndrome may vary, with the mild cases simply involving an elevation deficit in adduction. More severe cases include a hypotropia in primary position, a chin-up position and a head turn toward the involved side. The forced duction test is always positive when you attempt to elevate the eye in adduction, and upward saccades in adduction are usually rapid. A modification of the forced duction test includes doing it with anterior traction of the globe followed by another forced duction test with depression of the globe into the orbit. These additional measures are best performed in the operating room. In Brown's syndrome, the restriction of elevation in adduction will increase with depression of the globe into the orbit and diminish with anterior traction.
Acquired Brown's syndrome has been reported with nodular inflammatory reactions related to adult and juvenile forms of rheumatoid arthritis, pansinusitis, focal metastasis to the superior oblique muscle, frontal sinus osteorna, after orbital trauma, maxillofacial or sinus surgery or a superior oblique tuck. The acquired variety tends to be associated with pain or tenderness in the area of the trochlea and an audible click when the patient attempts to elevate the affected globe, as the superior oblique tendon passes through the trochlea. Acquired cases have a much higher incidence of spontaneous recovery, especially with appropriate medical management of the underlying condition. The differential diagnosis of a Brown's-type pattern of abnormal ocular motility includes the inferior rectus fibrosis syndrome, blow-out fracture, supranuclear paralysis of monocular elevation, myasthenia gravis, "trochleitis," adherence syndromes and idiopathic, isolated paresis of the inferior oblique muscle.
Other Causes
Inferior rectus fibrosis is a rare, congenital occurrence in which the involved eye is unable to elevate above the midline. In primary gaze, there is a large hypotropia and often ptosis on the same side. The forced ductions test reveals marked restriction to upward rotation while vertical saccades are normal velocity. The condition is most often unilateral, although one or both inferior rectus muscles may be affected. CT scanning has been reported in some cases to show atrophy of the involved inferior rectus muscle. Coronal views of the orbit are best suited for this purpose.
Strabismus fixus or generalized fibrosis of the extraocular muscles is a condition in which the globe is markedly limited in all fields of rotation. This congenital abnormality is often associated with head movements which are used to change the position of gaze in the absence of free eye movement. Forced ductions are positive in all directions. Voluntary eye movements are generally too small to adequately evaluate saccadic velocity.
During exploratory or retinal detachment surgery, one or more extraocular muscles may be disinserted to facilitate exposure of the diseased area. With retinal detachment surgery, a silicone sponge, buckle and an encircling element are frequently left on the globe to assure a successful retinal reattachment. This hardware plus the tissue dissection necessary for accurate placement, creates adhesions between the muscles, globe, orbital fat, Tenon's connective tissue and the silicone elements. The strabismus which may follow detachment operations may be horizontal, vertical or both. Diplopia is frequently present if vision of 20/200 or better is present in the operated eye.
With longstanding ocular muscle paralysis the antagonist muscle may become contractured. Usually, there is a large deviation and this deviation persists over time. The antagonist muscle, no longer having to exert equal tension on the globe, becomes short, tight and fibrotic. Two classic examples of this phenomenon are the "fallen-eye" and "rising-eye" syndromes.
The fallen eye syndrome occurs in a patient with a longstanding superior oblique muscle palsy who habitually fixates with the paretic eye. It manifests as a unilateral superior oblique palsy presenting as a hypodeviation of the uninvolved eye that worsens on abduction. The explanation is clear if you consider the effects of a long-standing superior oblique palsy, with underaction of the superior oblique muscle, overaction of the ipsilateral inferior oblique, and the patient's desire to fixate with the paretic eye. For example, if the right superior oblique is weak and the right eye is used for fixation, the ipsilateral, antagonist right inferior oblique—and its yolk, the left superior rectus (don't forget Sherrington's law)—receive less innervational input. Thus, testing of ocular versions (movement of both eyes together) will reveal that the left eye fails to elevate fully in abduction. With time, contracture of the contralateral (left) inferior rectus muscle develops. Although the underlying defect is in the right superior oblique, when the ipsilateral eye is used for fixation, the contralateral eye "falls" during abduction, secondary to prolonged hypodeviation of the non-paretic eye.
Conversely, the rising-eye syndrome occurs in patients with a long-standing inferior oblique palsy. If a person with a chronic left inferior oblique palsy also prefers to fixate with the weak left eye, less innervation flows to the ipsilateral left superior oblique and to the contralateral yolk, right inferior rectus. With chronic subnormal innervation of the right inferior rectus and shortening and eventual fibrosis of the right superior rectus muscle, the right eye will "rise" during attempted abduction.
The orbital pseudotumor syndrome is an ideopathic inflammatory condition of orbital tissues that frequently produces an acute, painful restrictive ophthalmopathy. There may be isolated or concomitant involvement of the extraocular muscles, muscle tendons, orbital fat, lacrimal glands, cranial nerves and the scleral shell. These patients usually present with acute-onset unilateral eye pain, proptosis, chemosis and extraocular muscle restriction. The forced duction test is positive in almost all cases.
Diagnosing the orbital pseudotumor syndrome and differentiating it from other acute painful orbitopathies can be accomplished by standardized ophthalmic echography, CT or MRI scanning. All these studies capably show enlargement of the extraocular muscles. CT and MRI may show contrast enhancement of the scleral shell, destruction of orbital bone, and periorbital sinus involvement. Standardized ophthalmic echography can give very specific information regarding the histopathologic composition of the affected orbital tissues. In acute myositis, for example, the affected eye muscle(s) and tendon(s) are enlarged and low reflective, because of an acoustically homogeneous infiltrate of inflammatory cells that characterizes most cases of orbital pseudotumor.
I hope that this review has clarified the following issues: 1) That vertical diplopia can result from either weakness of the agonist extraocular muscle or tethering of the antagonist. 2) The need to determine fixational preference in every case, early in the physical examination. 3) That a palsy of binocular or monocular eye movement can be related to a supranuclear or infranuclear lesion. 4) The consideration of restrictive ophthalmopathy as a cause of limited eye movement. 5) That each of the examination techniques used to clinically evaluate vertical diplopia has advantages and pitfalls that can lead the unwary examiner astray.
Dr. Spector is in private practice. Contact him at
1. King WM, Fuchs AF. Reticular control of vertical saccadic eye movements by mesencephalic burst neurons. J Neurophysiol 1979;42:861-876.
2. Graybiel AM, Haitwig FA. Some afferent connections of the oculomotor complex in the cat: An experimental study with tracer techniques. Bra