In the last century, microbiology textbooks have expanded their scope to include chapters on viruses, and recently, prions. The word "prion" was coined by Stanley B. Prusiner, MD, of the
In the last two decades, bovine outbreaks of "mad cow disease" have been prominently reported, particularly in the
Peculiar Prions
Although researchers aren't entirely certain what purpose prions serve, they believe that they're normally involved in useful pathways. The cellular protein prion (PrPc) occurs naturally, is likely synthesized in neurons,6 and attaches to the cell membrane through a glycosylphosphatidylinositol anchor. It is easily digested by proteinase K.7 PrPc is found in many tissues, with especially high levels in the brain;6 proposed functions are varied and include copper metabolism, signal transduction, memory formation, neuronal differentiation and lymphocyte activation.8 It's not until something triggers a translational malfunction, which causes the prion to fold differently at the secondary and tertiary levels, that the prion becomes highly resistant to denaturation.9 PrPSC—the "SC" refers to "scrapie"—aggregates in nervous tissue to form amyloid plaques,10 resulting in dementia, ataxia, convulsions and death. There is no treatment.
Several TSEs have been recognized in mammals: scrapie in sheep; bovine and feline spongiform encephalopathies; chronic wasting disease in deer and elk; transmissible mink encephalopathy; and exotic ungulate encephalopathy in nyala, oryx and greater kudu. Recognized human forms include Kuru, fatal familial insomnia, Gerstmann-Sträussler-Scheinker syndrome and various forms of CJD.11 All TSEs are fatal, and involve progressively debilitating neurological symptoms. While the abnormal protein structure most commonly results from a translation malfunction, it can also be introduced orally and iatrogenically.12,13 The abnormal protein is not easily destroyed, and usual cooking and sterilization techniques leave it unscathed.11
Taboo Practices
Kuru was epidemic in 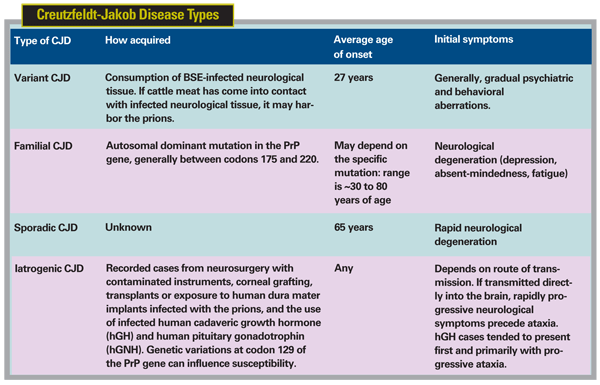
Similarly, bovine spongiform encephalopathy is spread through cattle herds by feed made with animal by-products.16 Only three cases of BSE have been reported in the
Various Presentations
Of course, not all cases of CJD can be traced to cows. There are four semi-distinct forms of the disease. Familial CJD is hereditary, has been traced to a mutations on the PRNP (prion protein) gene on chromosome 20, and accounts for 5 to 10 percent of CJD cases.20 The sporadic form (sCJD) appears without warning, generally in people over the age of 50, has a clinical duration of approximately four to five months, and accounts for approximately 85 percent of cases of CJD.20 These forms of CJD are not related to the consumption of BSE-contaminated meat. Iatrogenic CJD accounts for less than 5 percent of all CJD cases,20 and is the result of transmission of mutant prion (familial CJD, vCJD, or sCJD) via contaminated surgical equipment, transplantation of infected tissue or infusion of infected blood.
In 1990, following the BSE outbreak, the
As of January 2008, 166 definite or probable cases of vCJD have been reported in the U.K.23 Only three have been reported in the United States, and all have been linked to exposure during time spent in the U.K.24 Worldwide, the incidence of vCJD is approximately 0.5 to 1 per million, and all cases have been traced to the consumption of infected cattle meat from the U.K.25
There is concern, however, over the long incubation period and symptoms. All cases thus far have shown homozygosity at codon 129 of the PRNP gene—it may be that heterozygous individuals have a longer incubation period, and that the true size of the epidemic has yet to reveal itself.14,26
Iatrogenic Transfer
Although fewer incidences of vCJD have been reported since 2000, other forms of CJD, which can also be transferred iatrogenically, remain comparatively common.23 However, the risk of developing or contracting any form of CJD, especially outside of the
In 2003, the first cases of probable vCJD transmission through blood were reported.27,28 vCJD has also been shown to accumulate in the lymph, spleen and tonsils; therefore, the United States does not allow people who have resided for more than three months in a country where BSE is prevalent to donate blood.29 There has been one confirmed incident in which a cornea donor was found to have sCJD (diagnosed post-mortem and after the tissue donation) and the recipient later developed CJD; it occurred in the United States in 1974.13 At least two other possible transmissions of sCJD via corneal grafts have also occurred.30
Autopsies performed on sCJD and vCJD victims have shown no prion protein in any ocular structures other than the optic nerve and the retina. The retina seems to be particularly susceptible to prion-induced degeneration, although the degeneration is only clinically evident once the prions have replicated to maximal levels.31 In general, prions seem to be restricted to neurologic, immune-related and blood-fed tissues.21 Although no evidence exists of the transmission of CJD via ophthalmic instruments,32 during corneal transplants the blood-aqueous barrier is broken, compromising the immunologic privilege of the anterior chamber. If surgical instruments were previously contaminated, it is conceivable that prions could then be introduced to the patient. Hypothetically, the normal aqueous flow would wash any infectious agents out of the eye, and the prions should never come in contact with the retina or the optic nerve.
Concern has also been voiced over the transfer of epithelial cells on tonometers; again, no one has acquired CJD this way, but manual cleaning of the instruments to remove residual cells,33 the use of disposable instruments when appropriate and the use of cling film can help alleviate fears.34
Since 2004, the
As the number of confirmed vCJD cases has been decreasing, it's likely that the current agricultural regulations have halted its progression. Given the proposed extended incubation period, however, it's conceivable that more cases of vCJD will be revealed in the coming decades. Since iatrogenic transfer of CJD is not limited to vCJD, as the sporadic and familial forms can also be transmitted iatrogenically, reasonable caution is warranted to protect against this disease.
Current methods of tracing patients and medical instruments should be maintained, and the medical history of donors should be examined for symptoms indicative of vCJD. Recent research has shown soaking instruments in sodium hydroxide prior to autoclaving can significantly reduce prion levels in vitro;36 this should be considered when developing sterilization policies. Concern and precautions against vCJD ought to be particularly keen in the
In general, the use of disposable instruments for corneal surgery is preferable to prevent contamination of any kind, but the risk of vCJD transmission during corneal surgery appears to be minimal. Ultimately, the use of disposable instruments is an economic decision, left to the discretion of individual institutions.
Dr. Abelson, an associate clinical professor of ophthalmology at
1. Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982; 216:4542:136.
2. Wells GA, Scott AC,
3. Creutzfeldt HG. On a particular focal disease of the central nervous system (preliminary communication), 1920. Alzheimer Dis Assoc Disord 1989;3:1-2:3-25.
4. Hill A, Desbruslais M, Joiner S, et al. The same prion strain causes vCJD and BSE. Nature 1997;389:448-450.
5. Scott M, Will R, Ironside J, et al. Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proc Natl Acad Sci
6. Kretzschmar HA, Prusiner SB, Stowring LE, DeArmond SJ. Scrapie prion proteins are synthesized in neurons. Am J Pahtol 1986;122:1-5.
7. Oesch B, Westaway D, Wälchi M, et al. A cellular gene encodes scrapie Prp 27-30 protein. Cell 1985;40:4:735-46.
8. Hu W, Rosenberg RN, Stüve O. Prion proteins: a biological role beyond prion diseases. Act Neurol Scand 2007;116:75-82.
9. Pan KM, Baldwiin M, Nguyen J, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci 1993;90:10962-6.
10. Prusiner SB, Barry RA, McKinley MP, et al. Scrapie and Creutzfeldt-Jakob disease prions. Microbiol Sci 1985;2:2:33-9.
11. Vana K, Zuber C, Nikles D, Weiss S. Novel aspects of prions, their receptor molecules, and innovative approaches for TSE therapy. Cellular and Molecular Neurobiology 2007;27:1:107-28.
12. Prusiner SB, Cochran SP, Alpers MP. Transmission of scrapie in hamsters. J Infect Dis. 1985;152:5:971-8.
13. Duffy P, Wolf J, Collins G, et al. Possible person-to-person transmission of Creutzfeldt-Jakob disease. Letter to the editor. The
14. Collinge J, Whitfield J, McKintosh E, et al. Kuru in the 21st century – an acquired human prion disease with very long incubation periods. Lancet 2006;367:2068-74.
15. Prusiner SB, Gajdusek C, Alpers MP. Kuru with incubation periods exceeding two decades. Ann Neurol 1982;12:1:1-9.
16. Anderson RM,
17. Number of reported cases of bovine spongiform encephalopathy (BSE) in farmed cattle worldwide (excluding the
18. Restrictions on the importation of ruminants, meat and meat products from ruminants, and certain other ruminant products.
19. Onodera T, Kim CK. BSE situation and establishment of Food Safety Commission in
20. Variant Creutzfeldt-Jakob disease. World Health Organization Fact Sheet, revised November 2002. Accessed December 8, 2006. Available at http://www.who.int/mediacentre/factsheets/fs180/en/print.html
21. Wadsworth J, Joiner S, Hill A, et al. Tissue distribution of protease resistant prion protein in variant Creutzfeldt-Jakob disease using a highly sensitive immunoblotting assay. Lancet 2001;358:171.
22. Will R, Ironside J. A new variant of Creutzfeldt-Jakob disease in the
23. CJD Statistics. Provided by The National Creutzfeldt-Jakob Disease Surveillance Unit. Available at: http://www.cjd.ed.ac.uk/figures.htm. Accessed January 24, 2008.
24. Variant Creutzfeldt-Jakob Disease. Current Data Provided by the National Creutzfeldt-Jakob Disease Surveillance Unit. Available at:http://www.cjd.ed.ac.uk/vcjdworld.htm. Accessed January 25, 2008.
25. Introduction to Variant Creutzfeldt-Jakob Disease. Provided by the National Creutzfeldt-Jakob Disease Surveillance Unit. Available at http://www.cjd.ed.ac.uk/intro.htm. Accessed January 25, 2008.
26. Collee J, Bradley R, Liberski P. Variant CJD (vCJD) and bovine spongiform encephalopathy (BSE): 10 and 20 years on: part 2. Folia Neuropathol 2006;44:2:102-110.
27. Peden A, Head M, Ritchie D, Belle J, Ironside J. Preclinical vCJD after blood transfusion in a PRNP codon 120 heterozygous patient. Lancet 2004;264:527-529.
28. Llewelyn C, Hewitt P, Knight R, et al. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet 2004;363:417-421.
29. Guidance for Industry: Eligibility Determination for Donors of Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/Ps). Issued by the
30. Tullo AB, Buckley RJ, Kelly T, et al. Transplantation of ocular tissue from a donor with sporadic Creutzfeldt-Jakob disease. Clin Exp Ophthalmol 2006;34:645-9.
31. Hogan RN, Bowman KA, Baringer JR, Prusiner SB. Replication of scrapie prions in hamster eyes precedes retinal degeneration. Ophthalmic Res 1986;18:4:230-5.
32. Juan P, Ward H, De Silva R, Knight R, and Will R. Ophthalmic surgery and Creutzfeldt-Jakob disease. Br J Ophthalmol 2004;88:446-449.
33. Lim R, Dhillon B, Kurian K, et al. Retention of corneal epithelial cells following Goldman tonometry: implications for CJD risk. Br. J. Ophthalmol 2003;87:5:583-586.
34. Rani A, Dunne M, Barnes D. Cling film as a barrier against CJD in corneal contact A-scan ultrasonography. Ophthalmic and Physiological Optics 2003;23:1:9-12.
35. Creutzfeltdt-Jakob disease (CJD) and ophthalmology.
36. Bauman P, Lawrence L, Biesert L, et al. Critical factors influencing prion inactivation by sodium hydroxide. Vox Sanguinis 2006;91:34-40.


