The recently awarded Nobel Prize in chemistry recognized the contributions of two exceptional scientists, Robert Leftkowitz, MD, of Duke University and Brian Kobilka, MD, from Stanford University, for their work on the structure and function of g-protein coupled receptors, or GPCRs. This got us thinking about how important this family of signaling molecules is to ophthalmology, as they are to all areas of medicine. From Advair to Abilify, latanaprost to Lastacaft, and Vicodin to Viagra, GPCR-targeted drugs have transformed therapy for countless conditions. This month we'll consider the stuff from which scientific fame springs, taking a refresher course in g-protein signaling and a brief tour of some noteworthy GPCRs of the visual system.
Spotlight on G-proteins
The importance of g-proteins and the cell surface receptors that control their function is certainly reflected in the many scientists who've been honored by the Nobel committee.1 Julius Axelrod, Ulf von Euler, PhD, and Earl Sutherland, MD, won Nobel awards in the early 1970s for their studies of epinephrine and norepinephrine action, although many of these studies predate the concept of a g-protein. Alfred Gilman, PhD, and Martin Rodbell, PhD, shared the 1994 Nobel Prize for research which established the fundamentals of g-protein signaling, and Richard Axel, MD, and Linda Buck, PhD, were awarded the 2004 Prize for their work on g-proteins in the olfactory system. This level of attention from the Nobel committee is not surprising given that the gene family encoding GPCRs represents 1 percent of the human genome. The therapeutic impact may be even more impressive: It's been estimated that as many as half of all currently marketed drugs target some aspect of GPCR-based signaling pathways.2
Drs. Leftkowitz and Kobilka's research provides a clear, conceptual understanding of how hormones, neurotransmitters and drugs interact with these receptors and how those interactions lead to a remarkable diversity of responses seen throughout the body. As a fellow in Dr. Leftkowitz's lab in the late 1980s, Dr. Kobilka was an integral part of the team that first cloned adrenergic receptors and showed they were close relatives to rhodopsin.3 Since then, assembling and defining the many other participants in GPCR signaling has proceeded at a continually accelerating pace. While the Leftkowitz lab has focused on the intricate details of GPCR function,4 Dr. Kobilka's laboratory took on the herculean task of crystallizing purified samples of receptor protein.5 Protein crystals can be used to generate high-resolution structures which can provide a 3-D explanation for decades of biochemical studies. Using the analogy of the lock and key, most historical drug design has involved random testing to find keys that fit the lock of their target. The pairing of refined functional studies and crystallography of GPCRs, however, provides us with a dynamic model of the lock, and the opportunity to "cut keys" in a whole new way.
The Layers of GPCR Signaling
The function of any receptor is to detect chemical or physical messages in the extracellular world and translate those incoming messages into a coherent cellular response. GPCRs are positioned to do this as trans-membrane proteins, with one face exposed on the surface and another on the inside of the cell. The receptors cycle between three states that depend upon presence of an extracellular message, as depicted in Figure 1. That message can be a neurotransmitter, a hormone, a divalent ion or a photon.
|
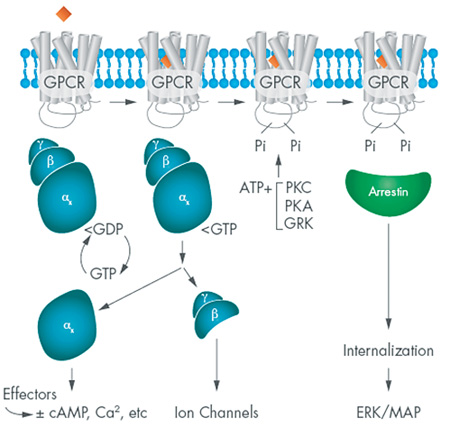
Although there are hundreds of different GPCRs, each typically couples with one of a dozen or so α subunits: thus, the beta adrenergic receptors commonly signal via an αsβϒ g-protein, while the histamine H1 receptor couples to αqβϒ heterotrimer. Each of these α subunits elicits a characteristic cellular response that may include modulation of adenylate cyclases, guanylate cyclases, or phospholipases. In addition, the βϒ dimer also participates in signaling in some cells by modulating the activity of membrane ion channels.
GPCR signaling can also be targeted by a receptor-independent g-protein modulation, and these are increasingly being recognized as physiological regulators of GPCR signaling.7,8 The best examples are the RGS proteins (regulators of g-protein signaling) that function to stimulate the GDP-GTP turnover independent of GPCR activation. Evidence for such proteins comes from many studies, including analysis of the GTPase activity of transducin, the heterotrimeric g-protein that partners with rhodopsin. Purified transducin hydrolyzes GTP to GDP with a t1/2 of about 15 seconds, but the rate of retinal deactivation has a t1/2 of about 0.5 sec.9 The human retinal RGS responsible for this effect is now designated RGS9, one of about two dozen such regulatory factors.10
It seems that all g-proteins have an intrinsic rate of GDP-GTP turnover, and the activation of GPCRs by ligands (or drugs) enhances that rate to trigger g-protein signaling. Overlaying this pathway are the RGS proteins, which act by further enhancement of GDP-GTP turnover. Of the 20 or so identified RGS proteins, most are specific for a single a subunit. Their role in modulating muscle contractility and maturation is well-established, and recent work suggests an important function in the immune system. There are also examples of RGS proteins in the visual system, and the interest in their functional importance and potential therapeutic targeting is likely to expand in the future.
GPCRs in the Eye
The majority of drugs used by practicing ophthalmologists, particularly those used to modulate autonomic function, act at GPCRs. The adrenergic receptors of the ciliary epithelium provide a clear example of how different g-protein α subunits and different GPCRs can orchestrate homeostatic control. Beta adrenergic GPCRs in the ciliary epithelium couple to a heteromeric g-protein with an αs subunit; activation of this receptor by epinephrine or other beta agonists leads to an increase in cellular cyclic AMP, which stimulates the secretion of aqueous humor. These same tissues also express alpha adrenergic receptors that couple to an αi subunit.11 The activation of this pathway causes an inhibition of cAMP synthesis and a reduction in aqueous humor. The net output will depend on the relative amounts of epinephrine and norepinephrine in the extracellular environment and their integration over time. Of course, both of these pathways are exploited in therapies for primary open-angle glaucoma, with beta antagonists such as timolol and alpha-2 agonists like brimonidine acting to suppress aqueous humor formation.
One ocular tissue where RGS proteins may impact GPCR function is the mast cells of the conjunctiva that underlie allergic conjunctivitis.12 Typically, mast cells are stimulated to release histamine via binding of surface IgE-antigen complexes, but degranulation can also be stimulated or potentiated by activation of GPCRs for adenosine (the A3 isoform) or chemokines such as CXCL12. These receptor pathways can also stimulate release of other vasoactive mediators such as IL-8, and they are regulated by RGS13. Surprisingly, RGS13 appears to have a function independent of its effect on GDP-GTP turnover, and over-expression of RGS13 in mast cells inhibits IgE-mediated degranulation, but this effect doesn't appear to be mediated via a heteromeric g-protein.13 Consistent with these findings, RGS13 knockout mice are hyper-responsive to IgE degranulation and more susceptible to anaphylaxis.13 Thus, RGS stabilizers or surrogates may find a use in treatment of allergic disease.
The classic example of a GPCR in the eye is rhodopsin, the visual receptor that responds to photons of light in much the same way as an adrenergic receptor does to epinephrine. The g-protein coupled to rhodopsin is called transducin, and it acts by shutting off a membrane current when it's in the active, GTP-bound phase of the cycle. Once the GTP is hydrolyzed, the transducin is deactivated, and the current is reactivated. This turns out to be a critical step in photo-accommodation, the process of visual adaptation when going from an area of bright light to darkness (or vice versa). For most people, this requires a few seconds to complete.
At the cellular level, photo-accommodation depends upon a retinal RGS protein, RGS9, that regulates the speed of transducin GDP-GTP turnover.14 In the past decade, researchers have identified a number of patients with bradyopsia, or slow vision, who require many minutes to adapt to changes in light intensity.15 In each case, this condition has been traced to mutations in either RGS9 or a related protein, R9AP, that acts to stabilize RGS9 in retinal cells. While this research was the first example of a human disorder linked to an RGS defect, it is not likely to be the last.
Future GPCR Discovery
The work of Drs. Leftkowitz and Kobilka (and others) has revealed the stunning complexity of GPCR signaling, and demonstrated a seemingly inexhaustible supply of opportunities for therapeutic intervention. Beyond simply targeting a molecule or a pair of interacting proteins for high-throughput-based approaches, we begin to more fully understand how we want our drugs of the future to interface with specific GPCR signaling pathways. For example, Dr. Leftkowitz's lab has identified and characterized a family of GPCR-interacting proteins, the arrestins, that were initially thought to be part of the cell's GPCR desensitization/recycling system.16 It turns out they do much more than that: Internalized GPCR-arrestin complexes bind with other cellular partners and activate a number of signal pathways, including the ERK/MAP pathway. Perhaps more interesting from a therapeutic perspective is that the degree to which arrestin pathways are activated is ligand-dependent, and may represent an underlying explanation for side effects associated with some GPCR targeted drugs. While this field is very young, it's thought that developing new ligands with a defined bias for activation of this second GPCR signaling pathway could find many therapeutic applications.
Pharmacologists love the idea of rational drug design but, unfortunately, as with many quests, the structural resolution of multiple GPCRs alone hasn't brought us to the end of the drug development rainbow. The power of Dr. Kobilka's structures is that he has paired them with functional studies that allow us to visualize the protein as it proceeds from resting to activated states.17 This is why their work is so important: Watching the movements of a complex protein such as a GPCR on an atomic scale will allow for dynamic modeling of drug binding sites and the transitions they undergo. Ultimately, knowledge that we gain from these observations will help us choose any phase of the GPCR activation pathway as a drug target. The therapeutic potential of such a choice seems eminently worthy of a Nobel Prize. REVIEW
Dr. Abelson is a clinical professor of ophthalmology at Harvard Medical School and senior clinical scientist at the Schepens Eye Research Institute. Dr. McLaughlin is a medical writer at Ora Inc., in Andover.
1. http://www.nobelprize.org/nobel_prizes/lists/ accessed 27 December 2012.
2. Kenakin T. New bull's-eyes for drugs. Sci American 2005;293:50-57.
3. Lefkowitz RJ, Kobilka BK, Benovic JL, et al. Molecular biology of adrenergic receptors. Cold Spring Harb Symp Quant Biol 1988;53:507-14.
4. Shukla AK, Xiao K, Lefkowitz RJ. Emerging paradigms of b-arrestin dependent seven transmembrane receptor signaling. Trends in Biochem Sci 2011;36:9:457-469.
5. Rasmussen SG, DeVree BT, Zou Y, et al. Crystal structure of the a2 adrenergic receptorGs protein complex. Nature 2011;477:549-557.
6. Gilman, A. G proteins: Transducers of receptor-generated signals. Annu Rev Biochem 1987;56:615649.
7. Kimple AJ, Bosch DE, Giguère PM, et al. Regulators of G-Protein Signaling and Their Ga Substrates: Promises and Challenges in Their Use as Drug Discovery Targets. Pharmacol Rev 63:728749, 2011.
8. Sjögren B, Neubig RR. Thinking outside of the ³RGS box²: New approaches to therapeutic targeting of regulators of G protein signaling. Mol Pharmacol 2010;78:4:550-7.
9. Vuong TM and Chabre M. Deactivation kinetics of the transduction cascade of vision. Proc Natl Acad Sci USA 1991;88:98139817.
10. Natochin M, Granovsky AE, Artemyev NO. Regulation of transducin GTPase activity by human retinal RGS. J Biol Chem 1997;272:28:17444-9.
11. Woldemussie E, Wijono M, Pow D. Localization of alpha 2 receptors in ocular tissues. Vis Neurosci 2007;24:5:745-56.
12. Bansal G, DiVietro JA, Kuehn HS, et al. RGS13 Controls G Protein-Coupled Receptor-Evoked Responses of Human Mast Cells. J Immunol 2008;181:7882-7890.
13. Bansal G, Xie Z, Rao S, et al. Suppression of immunoglobulin Emediated allergic responses by regulator of G protein signaling 13. Nat Immunol 2008;9:1:73-80.
14. Nishiguchi KM, Sandberg MA, Kooijman AC, et al. Defects in RGS9 or its anchor protein R9AP in patients with slow photoreceptor deactivation. Nature 2004;427:75-78.
15. Hartong DT, Pott JR, Kooijman AC. Six patients with Bradyopsia (slow vision). Ophthalmol 2007;114:2323-2331.
16. Whalen EJ, Rajagopal S, Leftkowitz RJ. Therapeutic potential of b-arrestin and GPCR biased agonists. Trends Mol Med 2011;17:126-139.
17. Shoichet BK, Kobilka BK. Structure-based drug screening for G-protein coupled receptors. Trends Pharm Sci 2012;33:268-272.


