Presentation
A 10-year-old Caucasian boy was referred to the Wills Eye Institute Pediatric Ophthalmology and Ocular Genetics service for the evaluation of a left recurrent retinal detachment. The patient was found to be myopic at 6 years of age. Two months prior to presentation, a routine examination revealed a chronic, shallow macula-off rhegmatogenous retinal detachment in the left eye. He was also found to have 180 degrees of lattice degeneration in the right eye. The patient underwent a scleral buckle procedure with cryotherapy and drainage of subretinal fluid in the left eye. In the right eye, prophylactic laser retinopexy was performed. One month later he had a recurrent macula-off retinal detachment with proliferative vitreoretinopathy. A pars plana vitrectomy with inferior retinotomy, endolaser and silicone oil placement was performed nine days prior to presentation.
Medical History
The patient reported no other medical problems. He was delivered full-term by cesarean section for breech presentation. Family history was significant for a maternal grandfather with a history of retinal detachment at age 12, who subsequently had bilateral retinal detachments and carries a diagnosis of Wagner syndrome. The patient's first cousin (son of his maternal aunt) was known to be myopic and also had a retinal detachment. The patient's mother was born with a cleft palate which had been attributed to medications taken by her mother during pregnancy. The patient's paternal grandmother had a retinal detachment at the age of 60.
Examination
Visual acuity in the patient's current spectacles (OD: -6.00 + 1.75 x 85, OS: -5.25 + 0.50 x 85) was 20/25 OD and counting fingers at three feet OS. His right pupil was 4 mm and reactive, and the left pupil was 7 mm and non-reactive with a 1+ left afferent pupillary defect. Extraocular motility was full. There was no strabismus. Intraocular pressure by applanation tonometry was 12 OD and 8 OS. Anterior segment examination of the right eye was unremarkable. The left eye showed 1+ cell and flare in the anterior chamber.
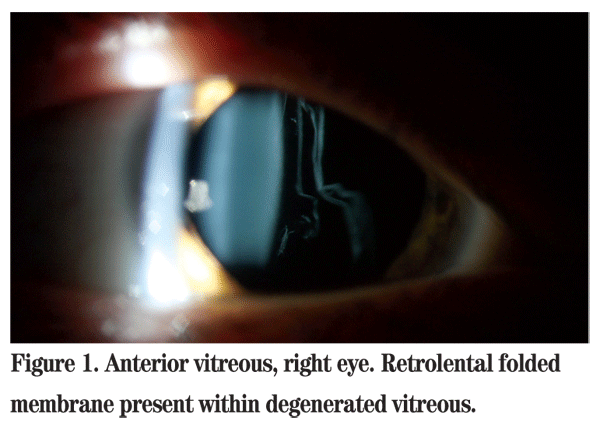
Examination of the anterior vitreous of the right eye revealed a folded membrane-like configuration of the vitreous in an otherwise optically empty vitreous cavity (See Figure 1). The previously noted areas of lattice degeneration and laser retinopexy were not easily visualized. Examination of the left eye showed silicone oil in the vitreous cavity. The retina appeared flat. Evidence of a 360-degree scleral buckle, recently applied cryotherapy and endolaser was apparent.
Diagnosis, Workup and Treatment
The differential diagnosis of an otherwise healthy young male with a retinal detachment, abnormal vitreous and a positive family history is the group of disorders known as the hereditary vitreoretinopathies including Wagner, Stickler and
Given the membranous appearance of the vitreous, history of retinal detachment, family history and physical examination, the patient's clinical picture was most consistent with type 1 Stickler syndrome. His first cousin had sequencing of the COL2A1 gene and was found to have a known pathogenic mutation, which confirmed the diagnosis. The patient was referred for genetic testing.
The patient's maternal grandfather who had been previously diagnosed with Wagner syndrome likely had Stickler syndrome with predominant ocular signs as is often times the case. By definition, the patient's mother was also affected, but she may be low expressing given her cleft palate. An eye examination will be conducted. Likewise, his maternal aunt was by definition affected and she too will be examined. One of the primary reasons for such evaluations of family members is to identify retinal abnormalities that may benefit from prophylactic laser photocoagulation. Genetic counseling can also be offered.
Stickler syndrome is a progressive arthro-ophthalmopathy and represents the most common cause of inherited rhegmatogenous retinal detachment in childhood.1,2 It has been subclassified into three types (STL1, STL2 and STL3) representing known mutated genes with autosomal dominant inheritance.2,3 Additional loci are also suspected.
STL1 and STL2 may be differentiated clinically based upon vitreous phenotype. The type 1 phenotype involves a mutation in the COL2A1 gene found on chromosome 12q13 which encodes for type II procollagen. Patients classically have retrolental membranous vitreous in an otherwise optically empty vitreous cavity. Type 2 arises as a result of mutation in the COL11A1 gene on chromosome 1p21, which encodes for the a1 chain of type XI procollagen. The vitreous in these patients has been described to have a fibrillar, beaded appearance. There is a non-ocular form of Stickler Syndrome, STL3, which has been associated with a mutation in the COL11A2 gene on chromosome 6p21.3. This gene encodes for the a2 chain of type XI procollagen and is not expressed in the vitreous.
These patients, however, can display the same characteristic systemic features of Stickler syndrome as those with STL1 and STL2.2,3,4 Additionally, an autosomal recessive form of Stickler syndrome exists involving the COL9A1 gene on chromosome 6q13. These patients have a distinct vitreous phenotype characterized by syneresis, which resembles the appearance of an aged vitreous.5
Phenotypic heterogeneity is marked, with variable expression within and among pedigrees. There is a subset of patients who may present only with the ocular manifestations of Stickler syndrome. Alternative splicing of exon 2 of the COL2A1 gene results in two forms of type II collagen, type IIA and type IIB. The former is coded for by all 54 exons of the COL2A1 gene and is principally expressed in the vitreous. The latter is coded for by 53 exons of the COL2A1 gene, exon 2 having been spliced out, and is mainly expressed in cartilaginous tissue. Consequently, mutations in exon 2 are considered to produce this predominantly ocular phenotype.6
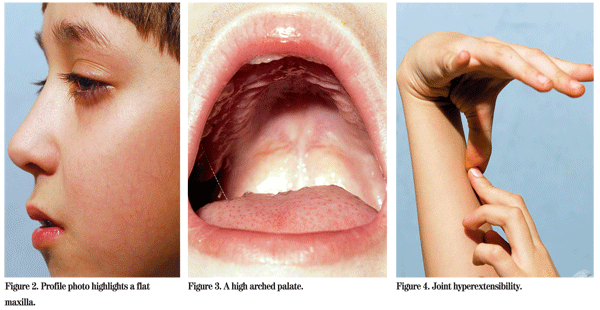
Other ocular manifestations of Stickler syndrome include axial myopia, cataracts (often cortical and wedge shaped), radial perivascular retinal degeneration and radial lattice degeneration. Rhegmatogenous retinal detachments are most often secondary to giant retinal tears. Non-ocular manifestations include midfacial hypoplasia, midline clefting ranging in severity from Pierre Robin sequence to bifid uvula, sensorineural hearing loss, joint hypermobility and premature osteoarthritis.2,4
Rhegmatogenous RD is the principal ocular complication of Stickler syndrome, reported to occur in up to 65 percent of patients.7 While many patients experience recurrent detachments following initial surgical management and require multiple procedures, good anatomical outcomes can be achieved with functional visual results.8 It has been postulated that RD secondary to Stickler syndrome be treated with primary vitrectomy and scleral buckle as a means of improving the primary success rate and achieving better clinical outcomes.8 In one large retrospective study, prophylactic 360-degree contiguous cryotherapy was an effective means of reducing the risk of RD in patients with type 1 Stickler syndrome.9 Today, vitreoretinal specialists use laser photocoagulation as a means of prophylaxis, however it is important to note that care should be taken during the laser procedure to avoid the long posterior ciliary nerves to minimize the chance of developing early presbyopia. Thus, the efficacy of prophylaxis makes early and correct diagnosis paramount to preserving vision.3
The author thanks Alex V. Levin, MD, MHSc of the Pediatric Ophthalmology and Ocular Genetics Service and Richard S. Kaiser, MD, of the Retina Service at Wills Eye Institute for their assistance with this manuscript.
1. Stickler GB, Belau PG, Farrell FJ, Jones JD, et al. Hereditary progressive arthro-ophthalmopathy. Mayo Clin Proc 1965;40:433-455.
2. Snead MP, Yates JR. Clinical and molecular genetics of Stickler syndrome. J Med Genet 1999;36: 353-359.
3. Edwards AO. Clinical features of congenital vitreoretinopathies. Eye 2008;22:1233-1242.
4. Niffenegger JH, Topping TM, Mukai S. Stickler's syndrome. Int Ophthalmol Clin 1993;33:271-280.
5. Van Camp G, Snoeckx RL, Hilgert N, van den Ende J, et al. A new autosomal recessive form of Stickler syndrome is caused by a mutation in the COL9A1 gene. Am J Hum Genet 2006;79:449-457.
6. Donoso LA, Edwards AO, Frost AT, Ritter R, et al. Clinical variability of Stickler syndrome: Role of exon 2 of the collagen COL2A1 gene. Surv Ophthalmol 2003;48:191-203.
7. Parma ES, Korkko J, Hagler WS, Ala-Kokko L. Radial perivascular retinal degeneration: a key to the clinical diagnosis of an ocular variant of Stickler syndrome with minimal or no systemic manifestations. Am J Ophthalmol 2002;134:728-734.
8. Abeysiri P, Bunce C, da Cruz L. Outcomes of surgery for retinal detachment in patients with Stickler syndrome: a comparison of two sequential 20-year cohorts. Graefe's Arch Clin Exp Ophthalmol 2007; 245:1633-1638.
9. Ang A, Poulson AV, Goodburn SF, Richards AJ, et al. Retinal detachment and prophylaxis in type 1 Stickler syndrome. Ophthalmology 2008;115:164-168.