Presentation
A 75-year-old Caucasian male presented to the Wills Emergency Room complaining of a painless decrease in vision in his right eye over a few days. He had a history of painless loss of vision in his left eye to no light perception three weeks prior for which he had been worked up at an outside hospital. His symptoms at that point had also included drooping of the left eyelid, a nonspecific headache and weight loss. At the outside hospital, the Westergren erythrocyte sedimentation rate (ESR), C-reactive protein (CRP) and platelet count were elevated. A temporal artery biopsy was performed and was negative for signs of giant cell arteritis. A head CT showed nonspecific swelling of the optic nerve on the left side. High-dose prednisone had been started at the previous hospital, and he was discharged on oral prednisone. He was transferred to the Wills Emergency Room because he was now losing vision in his right eye.
Medical History
The patient's past medical history was significant for osteoarthritis, diabetes, benign prostatic hypertrophy and skin carcinoma. He had been diagnosed with polymyalgia rheumatica a year prior, for which he received chronic, low-dose corticosteroid therapy. His past surgical history included Mohs' microsurgery for his skin carcinoma. The patient denied tobacco, alcohol or illicit drug use. His family history was non-contributory. On review of systems, he complained of malaise and chronic sinus congestion without epistaxis.
Examination
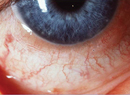
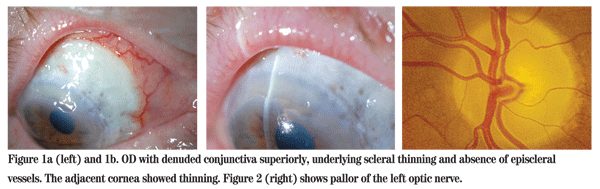
Visual acuity was 20/100 with 20/60 on pinhole OD, and no light perception OS. He had a frozen globe and ptosis on the left side. His extraocular motility was normal on the right and intraocular pressures were 12 mmHg OU. His left pupil was amaurotic, and the right pupil was reactive to light. The patient identified 13 of 15 Ishihara color plates OD. Anterior segment examination revealed areas of denuded conjunctiva superiorly OD with underlying scleral thinning and absence of episcleral vessels. The adjacent cornea was also thinned (See Figure 1). Dilated funduscopic exam was normal OD and revealed a pale disc OS (See Figure 2).
Diagnosis, Workup and Treatment
The differential diagnosis for a 75-year-old patient with sudden, painless vision loss with a history of polymyalgia rheumatica must include giant cell arteritis, which was appropriately worked up at the outside hospital. Progression of visual loss on corticosteroids may occur in GCA, and contralateral involvement despite appropriate corticosteroid doses is also possible. A negative temporal artery biopsy may occur in GCA for a variety of reasons, including inadequate length of specimen, blunted histopathologic findings secondary to chronic corticosteroid use, incorrect histopathologic diagnosis and discordance in temporal artery involvement (i.e., one side positive, contralateral side negative). Although isolated cranial neuropathy (usually abducens or oculomotor nerve palsy) may occur in GCA, the presence of a frozen globe makes this diagnosis less likely.
Our patient's clinical findings coupled with mild immunosuppression (e.g., chronic corticosteroid use, diabetes, renal disease, etc.) also raised the possibility of infection, including mucormycosis and aspergillosis. However, mucor is typically rapidly progressive and was lower on the differential diagnosis. Aspergillus infection remained a distinct possibility. The anterior segment findings suggestive of scleritis along with optic neuropathy and multiple cranial neuropathies are also seen in autoimmune disease, most notably Wegener's granulomatosis (WG).
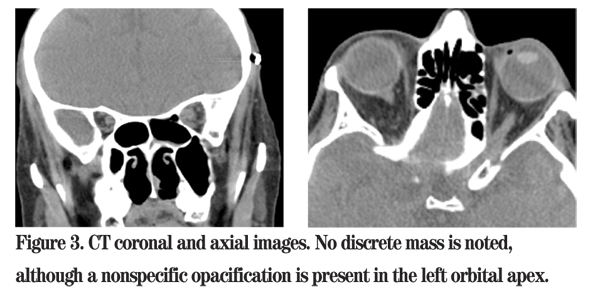
The patient was admitted to Wills Eye Institute with urgent consultations to Oculoplastics, Otolaryngology, Internal Medicine and Rheumatology.
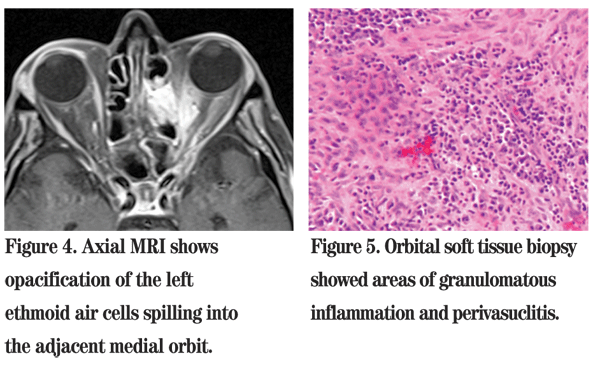
Chest X-ray showed normal lungs with no active disease. Given the patient's clinical scenario and imaging findings, an urgent transnasal endoscopic exploration of the left paranasal sinuses and orbital apex was performed using intraoperative image guidance. The histological findings on frozen section showed an area of perivasculitis suggestive of WG (See Figure 5). All specimens were negative for fungal elements.
The patient was started on cyclophosphamide 75 mg p.o. daily and methylprednisolone 250 mg q.i.d. for probable WG. The diagnosis was confirmed two days later with a positive c-ANCA titer of 1:80. At discharge nine days later, the patient's VA had improved to 20/50 OD and his scleritis had stopped progressing. Vision OS remained NLP.
Discussion
Wegener's granulomatosis is an uncommon, idiopathic, multisystem disorder characterized by small vessel granulomatous necrotizing vasculitis. Although the disease has a predilection for the upper and lower respiratory tracts and the kidney, clinical manifestations vary with the degree of organ involvement. Unexplained constitutional symptoms are often part of the initial presentation. Common clinical signs include chronic sinusitis, nasal septal perforation, "mulberry" gingivitis, hemoptysis, lung nodules, necrotizing glomerulonephritis, arthritis, cutaneous vasculitis and bullae, and polyneuritis. A more localized, sinoorbital variant of WG may also occur, with few or no associated systemic signs.
The incidence of WG is three per 100,000 persons, typically occurring during the fifth decade. There is no gender predilection, but Caucasians are affected more commonly than other races. It is likely that the prevalence of WG has been underestimated, since mild and indolent disease may go unrecognized for months to years.
The laboratory abnormalities associated with WG include anemia, leukocytosis, thrombocytosis, elevated erythrocyte sedimentation rate and elevated serum creatinine. Due to frequent renal involvement, urinalysis may reveal hematuria, proteinuria and RBC casts. The laboratory values in this patient were consistent with WG, and his elevated serum urea and creatinine likely indicated early renal involvement.
While thyroid eye disease is the most common orbital inflammatory disease, WG is the most common rheumatologic disease to involve the eye. Ocular manifestations reportedly occur in 28 percent to 58 percent of patients with WG and may be part of the initial presentation in 8 percent to 16 percent of cases. Ocular involvement ranges from mild conjunctivitis and episcleritis to severe scleritis, keratitis, uveitis and optic neuritis. Retinal vessel occlusion, nasolacrimal duct obstruction and retrobulbar involvement mimicking idiopathic orbital inflammation with proptosis have also been reported.1,2 Although the prevalence of scleritis is low in the general population (six per 100,000 persons), WG is high on the differential, as scleritis is seen in 7 percent of patients with WG.3 When WG is localized to the paranasal sinuses, eye and periocular tissue in the absence of systemic involvement, local tissue biopsy may be required for diagnosis.
WG belongs to a group of ANCA-associated vasculitides that also includes Churg-Strauss syndrome and microscopic polyangiitis. Various studies have showed the sensitivity of c-ANCA is over 90 percent in active systemic WG. However, in WG limited to the paranasal sinuses and orbits the sensitivity of c-ANCA can be as low as 32 percent.4 A recent retrospective case series studied the clinical utility of ANCA testing in predicting idiopathic scleritis patients who were at risk for developing WG. The results showed high specificity of ANCA testing for the development of WG in patients who present initially with isolated signs of idiopathic scleritis.5 Despite its excellent diagnostic value, quantitative measures of c-ANCA correlate erratically with disease activity; thus ANCA titers do not predict disease flares in a timely manner.6
WG ocular involvement will not respond to topical agents, except for some cases of limited anterior inflammation. Systemic anti-inflammatory and immunosuppressive therapy is the standard: prednisone (60 to 80 mg/day) and cyclophosphamide (2 mg/kg), with prednisone tapered after the cyclophosphamide has had time to take effect. In cases of cyclophosphamide intolerance, other agents, such as methotrexate, azathioprine or mycophenolate mofetil, may be used.3 The management of WG is challenging and a multidisciplinary approach is key to optimal care. Our patient remains stable visually, but flare-ups of his scleritis continue.
The authors would like to thank Jurij R. Bilyk, MD, of the Oculoplastics Service , and Mark L. Moster, MD, of the Neuro-Ophthalmology Service, at Wills Eye Institute, for their assistance with this case.
1. Harper SL, Letko E, Samson CM, et al. Wegener's granulomatosis: The relationship between ocular and systemic disease. J Rheumatol 2001;28(5):1025-32.
2. Pakrou N, Selva D, Leibovitch I. Wegener's granulomatosis: Ophthalmic manifestations and management. Semin Arthritis Rheum 2006;35(5):284-92.
3. Galor A, Thorne JE. Scleritis and peripheral ulcerative keratitis. Rheum Dis Clin North Am 2007;33(4):835-54.
4. Woo TL, Francis IC,
5. Lin P, Bhullar SS, Tessler HH, et al. Immunologic markers as potential predictors of systemic autoimmune disease in patients with idiopathic scleritis. Am J Ophthalmol 2008;145:463-471.
6. Klig JE. Ophthalmologic complications of systemic disease. Emerg Med Clin North Am 2008;26(1):217-31.