Diagnosis, Workup and Treatment
The patient, a 44-year-old immunocompetent Caucasian female with a diagnosis of MS secondary to presumed episodes of right eye optic neuritis, presented with 20/200 vision in the right eye and clinical findings of a unilateral occlusive arteritis, disc edema associated with peripapillary hemorrhages and macular edema.
|
Despite the patient’s prior diagnosis, multiple sclerosis was an unlikely cause for several reasons. First, MS causes a periphlebitis, not an arteritis.1 Approximately 11 percent of MS patients have venous sheathing on routine color fundus photography.2 Second, according to the Optic Neuritis Treatment Trial, optic disc edema associated with peripapillary hemorrhages was never found to lead to a diagnosis of MS. Finally, multiple sclerosis does not cause macular edema.
An extensive laboratory workup including anti-nuclear antibody (ANA), angiotensin converting enzyme (ACE), ANCA, RPR/FTA, PPD, Bartonella antibodies, rheumatoid factor, and HIV antibodies were all normal. The ESR was 17 mm/hr and CRP was slightly elevated at 1.7 mg/L. A chest X-ray was normal. While the laboratory tests were pending, the patient was admitted to the ophthalmology inpatient service and started on 1g intravenous methylprenisolone daily for three days. Given the retinal vasculitis and possible history of MS, a magnetic resonance imaging and magnetic resonance angiography of the brain were ordered. Imaging revealed non-specific scattered subcortical and periventricular white matter lesions on FLAIR imaging and no signs of cerebral vasculitis.
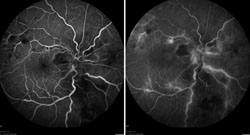
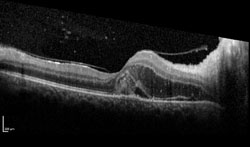
Fluorescein angiography helped confirm the findings of an occlusive arteritis (See Figure 2). The absence of aneurysms on FA helped ruled out IRVAN. Optical coherence tomography demonstrated macular edema and subretinal fluid seen on clinical exam (See Figure 3). HLA typing revealed our patient was HLA-B51 positive. Dermatology was consulted to administer a pathergy test, which was negative. In addition, a full exam of her skin and mucous membranes did not show any evidence of oral or genital sores. Despite the lack of additional findings, the leading diagnosis was possible Behçet’s disease.
Discussion
Diagnostic criteria for Behçet’s disease are clinical (See Table 1). The criteria were developed by the Behçet’s Research Committee of Japan in 1974 and were later revised in 2003 to give greater weight to the presence of ocular inflammation.3 Our patient only satisfied one major criterion.
Behçet’s disease is a chronic relapsing, idiopathic, multisystem, obliterative vasculitis that can affect both arteries and veins of all calibers. Although considered a multisystem disease, one system can preferentially be affected, leading to clinical sub-types such as ocular-Behçet’s disease. Theories exist that Behçet’s disease is an autoimmune condition triggered by an infectious agent in a genetically predisposed individual. The condition is most common in Eastern Mediterranean and Eastern-rim Asian populations, particularly along the Old Silk Road.4 The U.S. prevalence is 0.4 per 100,000 with a female predominance. Ocular involvement occurs in approximately 70 to 90 percent of cases and tends to affect both anterior and posterior structures.5 Eye involvement can be the initial presenting manifestation in 10 to 20 percent of cases. Anterior segment manifestations include a non-granulomatous anterior uveitis associated with a hypopyon, which shifts with gravity and head position. Posterior segment inflammation occurs in up to 93 percent of patients with ocular disease and portends a poor prognosis. Vascular occlusive episodes are a feared complication and if it involves the macula, visual acuity is reduced. Optic nerve involvement occurs in about 25 percent of patients. Classic dermatologic findings include oral aphthous ulcers, erythema nodosum, genital ulcer, and acne vulgaris.
|
The initial goal of therapy is swift control of intraocular inflammation, typically accomplished using a combination of high-dose intravenous, oral, periocular, intravitreal or topical corticosteroids. Equally important is preventing additional inflammatory episodes. Long-term treatment with steroids should be avoided in cases of significant posterior uveitis, as chronic steroid use has not been found to reduce recurrences or improve the visual prognosis.6 Many steroid-sparing agents currently exist to prevent recurrence of inflammation. Some of the most commonly employed steroid-sparing medications include infliximab, azathioprine, cyclosporine-A, cyclophosphamide, tacrolimus and methotrexate.
After three days of IV methylprednisolone, our patient was transitioned to oral prednisone. Over the next month, the inflammation and macular edema resolved. Her visual acuity returned to 20/30 with full recovery of her color vision (See Figures 4 & 5). Rheumatology did not recommend steroid-sparing agents and continued tapering her oral prednisone with no recurrence to date. REVIEW
The author would like to thank Robert C. Sergott, MD, of the Wills Eye Neuro-Ophthalmology Service, for his time and assistance in preparing this case.
1. Rucker WC. Sheathing of the retinal veins in Multiple Sclerosis. Mayo Clinc Proc 1972;47:335-40.
2. Young BR. Fluorescein angiography and retinal venous sheathing in multiple sclerosis. Can J Ophthalmology 1976;11:31-6.
3. Kurokawa MS, Suzuki N. Behçet’s disease. Clin Exp Med 2004;3:10-20.
4. Ohno S. Behçet’s Disease in the world. In: Recent Advances in Behçet’s disease, Lehnet T. Barnes C.G. (Eds) Royal Society of Medicine Service, London.
5. Verity DH, et al. Behçet’s disease, the Silk Road and HLA-B51: Historical and geographic perspectives. Tissue Antigens 1999;4:213-220.
6. Tugal-Tutkin I, et al. Uveitis in Behçet’s disease: An analysis of 880 patients. Am J Ophthalmol 2004;136:1114-9.


