Diagnosis, Workup and Treatment
From the clinical history and exam, a bilateral branch retinal artery occlusion was suspected secondary to sickle cell disease. The event was likely triggered by dehydration. A fluorescein angiogram demonstrated bilateral macular and peripheral focal non-perfusion (See Figures 4, b & c). Optical coherence tomography showed retinal atrophy and edema in both eyes.
The patient was admitted to the hospital with further treatment coordinated in association with her hematologist. Complete blood count was remarkable for low hemoglobin, and a transfusion of packed red blood cells as well as intravenous fluids was given. A further thrombophilia workup was deemed unnecessary, given the likely known etiology of sickle cell disease.
|
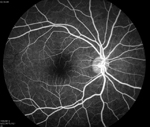
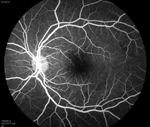
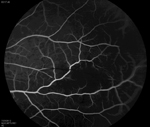
Discussion
Sickle-cell disease is an inherited autosomal recessive disease due to mutations in the beta-globin gene on the short arm of chromosome 11. The most common form, designated SS disease, occurs in individuals homozygous for a single point mutation that causes a substitution of valine for glutamic acid at the sixth position in the beta-globin chain. This point mutation results in the production of hemoglobin S. A less common form, SC disease, occurs when an individual has one copy of the allele for hemoglobin S, as well as an allele for hemoglobin C, in which a mutation causes a substitution of lysine for glutamic acid.
Worldwide, around a quarter of a million children are born each year with sickle-cell disease, about 60,000 of which are in the United States. The sickle-cell allele is much more common in African populations, or populations of African descent. Approximately 0.15 percent of all African-American children have SS disease.
Although ocular disease due to sickle cell is more prevalent in SC patients compared to SS patients, the various ocular manifestations of sickle-cell disease occur in both forms. Ocular findings occur in the anterior segment as well as the posterior segment. Comma-shaped capillary segments, most commonly seen on the inferior bulbar conjunctiva, may be present due to transient dilatation of conjunctival blood vessels by abnormally shaped red blood cells.1 The comma-shaped capillary segments often decrease under the heat of the slit-lamp beam as a result of vasodilation. Sectoral iris atrophy and pupillary irregularities can be seen when iris infarcts occur. At the disc, small dilated capillary vessels appear as small red dots in a linear or Y-shape pattern. These segments consist of pre-capillary arterioles occluded with sickled red blood cells.2
|
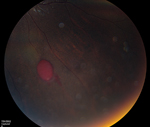
Proliferative sickle-cell retinopathy is the most vision-threatening complication of sickle-cell disease. It occurs at the junction of perfused and nonperfused retina, most commonly found in the superotemporal followed by the inferotemporal quadrants.6 Peak prevalence in SS patients is between 25 and 39 years, with no gender predilection, and in SC patients between 15 and 24 years in men and 20 to 39 years in women. The natural history of proliferative sickle-cell retinopathy begins with peripheral vascular occlusion causing local ischemia and production of vascular growth factors. This causes vascular remodeling and arteriovenous anastomoses, followed by retinal neovascularization, usually in a sea fan shape. The new retinal vessels can cause vitreous hemorrhage and tractional retinal detachments, or they may spontaneously regress. Around 21 to 23 percent of SC patients and 2 to 3 percent of SS patients will have retinal neovascularization with vitreous hemorrhage, and as much as 60 percent of sea fan neovascularization will resolve spontaneously via autoinfarction.7
Given the high rates of autoinfarction with spontaneous resolution of proliferative sickle-cell retinopathy, asymptomatic new blood vessels that are not macula-threatening can be observed. Previous treatment modalities have included feeder arteriolar occlusion and cryotherapy, but the current mainstay of treatment is laser photocoagulation. The role of anti-vascular endothelial growth factor agents is not yet clear. Tractional retinal detachments, nonclearing vitreous hemorrhage and macular holes may all be treated with vitrectomy if the vision is affected.
The author would like to thank Mike Dollin, MD, vitreoretinal fellow of the Wills Eye Retina Service, for his time and assistance preparing this case.
1. Paton, D. The conjunctival sign of sickle cell disease. Arch Ophthalmol 1961;66:90-4.
2. Serjeant GR. The clinical features of sickle cell disease. New York: Elsevier, 1974.
3. Goldbaum MH, Goldberg MF, Nagpal K, et al. Proliferative sickle retinopathy, in L’Esperance F. (ed). Current Diagnosis and Management of Chorioretinal Disease. St Louis: CV Mosby Co, 1976,132-145.
4. Fine LC, Petrovic V, Irvine AR, et al. Spontaneous central retinal artery occlusion in hemoglobin SC disease. Am J Ophthalmol 2000;130:680-1.
5. Gagliano DA, Goldberg MF. The evolution of salmon-patch hemorrhages in sickle cell retinopathy. Arch Ophthalmol. 1989;107:1814-5.
6. Fox PD, Dunn DT, Morris JS, et al. Risk factors for proliferative sickle retinopathy. Br J Ophthalmol 1990;74:172-6.
7. Downes SM, Hambleton IR, Chuange EL, et al. Incidence and natural history of proliferative sick cell retinopathy: Observations from a cohort study. Ophthalmology 2005;112:1869-75.


