Retinitis pigmentosa most often strikes younger patients, including those in the pediatric age group, and the onset of Leber’s congenital amaurosis, a disease associated with RP, can occur in infancy. For years, we could do nothing but chart the disease’s damage to our patients’ vision. With the Food and Drug Administration approval of Luxturna (voretigene neparvovec/AAV2-hRPE65v2; Spark Therapeutics, Philadelphia) late in 2017, however, we finally have a treatment for this disease.
This article will discuss the basic mechanism of gene therapy, the promising results of Luxturna’s trial, and the challenges that gene therapies like Luxturna must surmount in the future.
Etiology of RP
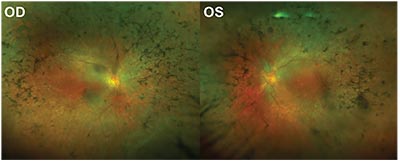 |
| Figure 1. Fundus images of a patient with retinitis pigmentosa. |
Three million people across the globe are afflicted with retinitis pigmentosa, a hereditary disease that causes progressive degeneration of the retina’s rod and cone cells that eventually leads to total or near-total blindness (See Figure 1). Unfortunately, given the disease’s devastating effects, there is often little that can be done to improve visual outcomes in these patients.
Part of the difficulty in treating RP is its complex and diverse genetic etiology: There are approximately 70 known genes and 3,000 genetic mutations associated with RP. Furthermore, RP is genetically associated with other retinal degenerative diseases, such as Leber’s. While there are several different supportive treatments available, including supplementation with vitamin A and omega-3 fatty acids, or usage of neurotrophic factors and antioxidants, these therapies have often been been shown to be ineffective, failing to address the root cause of the disease. However, gene therapy—a technique by which wild-type cDNA is delivered to the host retina’s genetically defective cells via a viral vector—has begun to show promising results for improving visual outcomes, as evidenced by new clinical trials like the one used to secure Luxturna’s approval.1,2
The RPE65 Gene Mutation
A variety of genetic mutations cause RP, but the sole target of Luxturna is the uncommon, autosomal recessive mutation of gene RPE65. Only about 5 to 20 percent of all forms of RP are autosomal recessive and, of those, only about 2 to 5 percent of all recessive forms of RP involve the RPE65 mutation.3 The genetic codes for the RPE65 enzyme, also called retinoid isomerohydrolase, are found within the cells of the retinal pigment epithelium. RPE65 normally regenerates 11-cis-retinal, a molecule essential to the opsins (visual pigments) of the photoreceptors (See Figure 2). When both copies of the RPE65 gene are defective, the RPE65 enzyme isn’t produced and visual pigment can’t be regenerated, leading to photoreceptor dysfunction and death.1
Patients with two copies of the autosomal recessive RPE65 mutation will develop complete blindness in both eyes at an early age. Depending on when the patient is diagnosed, the patient is deemed to have LCA or early onset RP. The effect of this mutation has been established through animal models, with studies of dog and mouse models with RPE65 knockout (RPE65–/–) demonstrating a disease progression that mimics the pattern of disease found in LCA or early-onset RP in humans.4,5,6
Early Trials of Gene Therapy
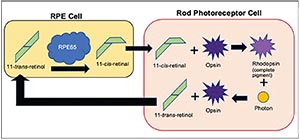 |
| Figure 2. Simplified schematic showing the role of the RPE65 enzyme (retinoid isomerohydrolase) in visual pigment restoration. Note that RPE65 is among several of the enzymes (not shown) in the RPE cell that participate in regenerating visual pigment. |
Gene therapy requires two main components: the non-mutated, wildtype version of the given DNA sequence encoding for a functional protein—in this case, RPE65 encoding for the functional RPE65 enzyme— and the delivery vector (See Figure 3). Previous investigations into retinal gene therapy have demonstrated that the adeno-associated virus (AAV) is be among the safest and most experimentally malleable vectors for delivery to deep retinal cells.7 The vector needs to be injected subretinally to reach its target, the RPE layer. Research has shown RPE cell transduction success with many serotypes of the AAV vector.8-10 Studies in animal models demonstrated that subretinal injection of RPE65 cDNA via an AAV vector into the RPE65 sequence enhanced visual acuity and ERG readings.11,12
Beginning in 2008, the first Phase I/II human clinical trials were performed using this model on 12 human patients with the autosomal recessive RPE65 mutation. However, the findings were mixed, with some patients showing only short-lived improvement in retinal function, and others showing no improvement or severe retinal decline.13,14 Other Phase I/II clinical trials were subsequently performed, but also didn’t show significant visual improvement.11,15-17
To address these outcomes, modifications to the RPE65 gene promoter and AAV2 vector apparatus were made in another Phase I/II clinical trial, sponsored by Spark Therapeutics. Twelve patients with the autosomal recessive RPE65 mutation were recruited for the trial. After two years of subretinal injections in one eye, all of the study patients demonstrated improvement in retinal function and visual acuity measurements in the injected eye without any major adverse incidents.18,19 A follow-up trial involving 11 of these 12 patients, in which the AAV2 vector was injected into each patient’s other eye showed improvement in retinal function across all patients that lasted for about three years.20
Luxturna is Born
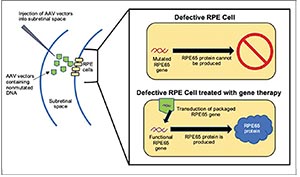 |
| Figure 3. Simplified schematic of the mechanism behind RPE65-targeted gene therapy. |
The promising results of Spark Therapeutics’ early trials led the research team in July 2017 to publish a randomized, open-label, Phase III clinical trial of Luxturna. A total of 29 patients with autosomal recessive RPE65 mutations participated in the study, with 20 individuals receiving the treatment and nine acting as controls. Luxturna was injected subretinally in both eyes of each patient in the intervention group. The efficacy of the intervention was determined via the Multi-Luminance Mobility Test score, measured at baseline prior to intervention and at follow-up one year later.
In the MLMT, patients are asked to navigate through an obstacle course at different environmental light levels. The MLMT assesses not only visual acuity, but also light perception, contrast sensitivity and visual field. At the one-year follow-up, the MLMT score in the intervention group improved by 1.8 (SD 1.1), whereas the MLMT score in the control group improved by only 0.2 (SD 1.0)—a statistically significant difference (p=0.0013). Furthermore, a large percentage of the intervention group (65 percent) navigated the MLMT successfully at the minimum luminance setting of 1 lux, compared to none of the controlgroup patients. There were no immunological reactions or other major adverse events.2 Spark Therapeutics’ Phase III trial thus demonstrated that the treatment is both safe and effective and, in December 2017, the FDA approved the use of Luxturna for individuals afflicted with retinal degenerative disease due to RPE65 mutation.21
Triumphs and Challenges
Luxturna casts a positive light on the outlook for the success of gene therapy to completely restore vision and vastly improve the quality of life of those with severe visual impairment from RP, LCA and other retinal degenerative diseases. In contrast to other treatment approaches, Luxturna’s mechanism specifically, and gene therapy in general, offer several key advantages.
First, injections into the retina will rarely elicit an immune response, due to the blood-retina barrier and the eye’s naturally immunosuppressive environment. Thus, gene therapy is a relatively safe means of treatment, and is likely to become even safer as the technology for vector delivery advances.7 Another advantage of gene therapy is that it addresses the fundamental etiology of the disease: the defective genes. Unlike artificial retinal implants—which are inserted only in the late stages of disease when most of the photoreceptor layer is already destroyed—gene therapy can be implemented in the earlier stages. Gene therapy therefore has potential to save the photoreceptors from an otherwise doomed genetic fate.19
However, there are still several important challenges for gene therapies like Luxturna. One major disadvantage of Luxturna is its severely limited therapeutic target, since it is effective only for the 1,000 to 2,000 patients in the United States with the recessive RPE65 mutation. Many more thousands of patients suffering from hereditary retinal degenerative disease do not yet have a viable gene therapy. Unfortunately, gene therapy tends to work best only for recessive and X-linked mutations, which result in complete failure of production of a functional, essential protein. In this situation, the nonmutated versions of the genes can be delivered to the necessary cells, replacing the mutated genes and allowing for normal production of the essential protein. Gene therapy doesn’t work as well for dominant mutations, which are the cause of 15 to 25 percent of RP cases, due to the “dominant negative” phenomenon.1 Dominant mutations result in the overproduction of a dysfunctional protein, negating the effect of any functional protein present. Therefore even if the gene for functional proteins were delivered to the cells, it’s unlikely that enough functional protein could be made to overcome the mass quantities of dysfunctional protein made by the patient’s own native genes.22-24
Another drawback of Luxturna is the need to inject the delivery vector into the subretinal space. Surgical invasion into the subretinal space necessitates inducing a temporary retinal detachment and increases the chance for necrosis and inflammation in the area. A potential solution to this problem is to deliver the vector into the vitreous humor, which avoids direct contact with the retina, and would also make the therapy more accessible for ophthalmologists who haven’t undergone vitreoretinal surgical training. Yet, previous trials with gene therapy delivered through intravitreal injection have shown diminished visual outcomes and higher chances of an immune response; for adequate vector-cell transduction to occur, the vectors need to be placed in close proximity to the deeper layers of the retina (the subretinal space). Future research in gene therapy should therefore focus upon ways to optimize vector delivery to render intravitreal injections more feasible.18,25,26
One last disadvantage of Luxturna is its current exorbitant cost of $425,000 per eye. Even though the product has just entered the market as a promising innovation, critics argue that it’s difficult to justify the price given that the long-term efficacy of the treatment is still unknown.27
Gene therapy is a new and developing field. We hope that as advancements in gene therapy are made and the field becomes more mainstream, treatment will become more affordable, accessible and effective. With every innovation like Luxturna that comes into the foreground, we move one step closer to curing retinal degenerative disease. REVIEW
Jonathan Barnett is an MD candidate in the Class of 2019 at Stony Brook School of Medicine in Long Island, N.Y. Dr. Landa is a practicing vitreoretinal surgeon at the New York Eye and Ear Infirmary of Mount Sinai and an associate professor of ophthalmology at Icahn Medical School of Mount Sinai. Neither author has a financial interest in any of the products discussed.
1. Dias M, Joo K, Kemp J, et al. Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Progress in retinal and eye research 2017;63;107131
2. Russell S, Bennett J, Wellman JA, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65mediated inherited retinal dystrophy: a randomized, controlled, open-label, phase 3 trial. The Lancet 2017;390;849-60.
3. Ferrari, S, Di Iorio E, et al. Retinitis pigmentosa: Genes and disease mechanisms. Curr Genomics 2011;12;238–249.
4. Cideciyan AV. Leber congenital amaurosis due to RPE65 mutations and its treatment with gene therapy. Prog Retin Eye Res 2010;29;398–427.
5. Nilsson SE, Wrigstad A, Narfstrom K. Changes in the DC electroretinogram in Briard dogs with hereditary congenital night blindness and partial day blindness. Exp Eye Res 1992;54;291– 296.
6. Rohrer B, Goletz P, et al. Correlation of regenerable opsin with rod ERG signal in Rpe65-/mice during development and aging. Invest Ophthalmol Vis Sci 2003;44;310–315.
7. Dalkara D, Goureau O, et al. Let there be light: Gene and cell therapy for blindness. Hum Gene Ther 2016;27;134–147.
8. Auricchio A, Kobinger G, et al. Exchange of surface proteins impacts on viral vector cellular specificity and transduction characteristics: The retina as a model. Hum Mol Genet 2001;10;3075–3081.
9. Dinculescu A, Glushakova L, et al. Adeno-associated virusvectored gene therapy for retinal disease. Hum Gene Ther 2005;16;649–663.
10. Weber M, Rabinowitz J, et al. Recombinant adenoassociated virus serotype 4 mediates unique and exclusive long-term transduction of retinal pigmented epithelium in rat, dog, and nonhuman primate after subretinal delivery. Mol Ther 2003;7;774–781.
11. Jacobson SG, Acland GM, et al. Safety of recombinant adeno-associated virus type 2-RPE65 vector delivered by ocular subretinal injection. Mol Ther 2006;13;1074–1084.
12. Bennicelli J, Wright JF, et al. Reversal of blindness in animal models of leber congenital amaurosis using optimized AAV2mediated gene transfer. Mol Ther 2008;16;458–465.
13. Bainbridge JW, Smith, AJ, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med 2008;358;2231–2239.
14. Bainbridge, JW, Mehat MS, et al. Long-term effect of gene therapy on Leber’s congenital amaurosis. N. Engl. J. Med 2015;372;1887–1897.
15. Hauswirth, WW, Aleman TS, et al. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: Short-term results of a phase I trial. Hum Gene Ther 2008;19;979–990.
16. Cideciyan AV, Hauswirth WW, et al. Human RPE65 gene therapy for Leber congenital amaurosis: Persistence of early visual improvements and safety at 1 year. Hum Gene Ther 2009;20;999–1004.
17. Jacobson SG, Cideciyan AV, et al. Improvement and decline in vision with gene therapy in childhood blindness. N Engl J Med 2015;372;1920–1926.
18. Maguire AM, Simonelli F, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med 2008;358;2240–2248.
19. Maguire AM, High KA, et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: A phase 1 doseescalation trial. Lancet 2009;374;1597–1605.
20. Bennett J, Wellman J, et al. Safety and durability of effect of contralateral-eye administration of AAV2 gene therapy in patients with childhoodonset blindness caused by RPE65 mutations: A follow-on phase 1 trial. Lancet 2016;388;661–672.
21. FDA approves novel gene therapy to treat patients with a rare form of inherited vision loss. U.S. Food and Drug Administration 2017 [cited March 2018]. Available from: https://www.fda.gov/ NewsEvents/Newsroom/PressAnnouncements/ucm589467.htm. 22. Dalkara D, Sahel JA. Gene therapy for inherited retinal degenerations. C R Biol 2014;337;185–192.
23. Chadderton N, Millington-Ward S, et al. Improved retinal function in a mouse model of dominant retinitis pigmentosa following AAV-delivered gene therapy. Mol Ther 2009;17;593– 599.
24. Petrs-Silva H, Linden R. Advances in gene therapy technologies to treat retinitis pigmentosa. Clin Ophthalmol 2014;8;127–136. 25. Dalkara D, Kolstad KD, et al. Inner limiting membrane barriers to AAV-mediated retinal transduction from the vitreous. Mol Ther 2009;17;2096–2102.
26. Dalkara D, Byrne LC, et al. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci Transl Med 2013;5;189.
27. Berkrot, B. Spark’s price for Luxturna blindness gene therapy too high: ICER. Reuters 2018 [cited March 2018]. Available from: https://www.reuters.com/article/us-spark-icer/sparks-price-forluxturna-blindness-gene-therapy-too-high-icer-idUSKBN1F1298.


