Great strides have been made over the last decade in therapeutic regimens for wet age-related macular degeneration. Modalities involving thermal or selective laser ablation, radiotherapy and surgical extraction of neovascular tissue have largely been replaced by targeted pharmacotherapy. Increased understanding of the molecular mechanisms involved in neovascularization has led to identification of relevant targets, such as vascular endothelial growth factor-A, an endothelial cell mitogen and survival factor. Prospective randomized clinical trials addressing the treatment of wet AMD have convincingly demonstrated the benefit of targeting VEGF.
 While pegaptanib (Macugen) was the first FDA approved anti-VEGF agent, its efficacy was limited with respect to patients gaining significant vision (three lines of visual gain using a standardized visual acuity chart). Subsequent clinical trials have demonstrated that other agents targeting VEGF, such as ranibizumab (Lucentis), bevacizumab (Avastin) and aflibercept (VEGF-Trap) result in significant visual gain in approximately one-third of the patients with wet AMD. (Nguyen Q, et al. IOVS 2011;52:ARVO E-Abstract 3128)1-3
While pegaptanib (Macugen) was the first FDA approved anti-VEGF agent, its efficacy was limited with respect to patients gaining significant vision (three lines of visual gain using a standardized visual acuity chart). Subsequent clinical trials have demonstrated that other agents targeting VEGF, such as ranibizumab (Lucentis), bevacizumab (Avastin) and aflibercept (VEGF-Trap) result in significant visual gain in approximately one-third of the patients with wet AMD. (Nguyen Q, et al. IOVS 2011;52:ARVO E-Abstract 3128)1-3
There is widespread acceptance that the majority of the therapeutic benefit of monotherapy anti-VEGF agents in use today is due to their potent anti-permeability property.4 Anti-VEGF monotherapy does not, however, induce disease-modifying regression of the pathologic NV tissue.1-3 Therefore, stability of the NV complex ensues despite continued treatment, which may limit significant visual gain in a majority of treated patients. Hence, targeted pharmacotherapeutic strategies inducing NV regression coupled with resolution of permeability in wet AMD may result in enhanced visual outcome.
Neovascularization is a normal process involved in embryonic development and tissue repair but becomes pathological when blood vessels proliferate into abnormal tissues (i.e., tumors), the avascular cornea or the subretinal space. The proliferation, invasion and migration of the NV vessels are coordinated by complex molecular mechanisms involving growth factors, cytokines and their associated receptors. The diverse set of signaling pathways sets the stage for a multi-targeted pharmacotherapy approach for the treatment of AMD.
The NV tissue consists of endothelial cells, pericytes and inflammatory cells (i.e., occasional macrophages). Based on our current understanding, the NV process involves the formation of angiogenic sprouts, led by tip cell filopodia, from existing capillaries into the avascular space. VEGF is a master switch responsible for activation, survival and proliferation of EC and has been found in the localized space at the tip of the angiogenic sprout. Following and lagging behind the EC angiogenic sprout formation, the newly formed vessels attract and are coated by pericytes, leading to NV maturation.
Pericytes are derived via differentiation of mural cells. Their presence on capillaries not only leads to NV support and stabilization but promotes endothelial cell survival through chemical signaling and physical interactions including pericyte production of VEGF.6 This endothelial survival signaling by integrated pericytes is critical and may explain the resistance of the NV tissue to VEGF withdrawal, i.e., lack of NV regression to monotherapy anti-VEGF treatment. Therefore, stripping of pericytes combined with anti-VEGF therapy may lead to regression of NV vessels and improved visual outcomes.
The coating of the NV endothelial cells by pericytes is initiated by endothelial cell expression of the paracrine platelet-derived growth factor B, which forms the homo dimer PDGF-BB. The EC expressed PDGF-B is retained proximally by heparin sulfate proteoglycan, with the highest concentration in the tip cells of the angiogenic sprout. The recognition of the proximally retained PDGF-BB by the pericyte-bound PDGFR-β receptor initiates the proliferation and migration of the pericytes along the growing neovascularization. The role of PDGF-B/PDGFR-β interaction for pericyte recruitment and capillary maturation has been demonstrated through gene knockout, ectopic delivery of competing PDGF-B, blocking the PDGF-B/PDGFR-β interaction by external agents, or deletion of the PDGF-B heparin sulfate proteoglycans binding motif.4 The attachment and maturation of the pericytes lags behind the endothelial cell proliferation at the angiogenic sprout in both time and space.7,8
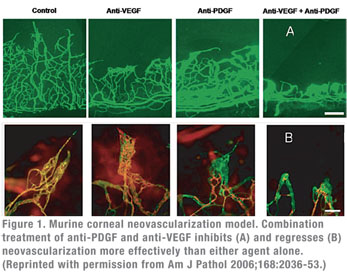
The importance of the PDGF-B/PDGFR-β interaction in pericyte recruitment, maturation and resistance to anti-VEGF mediated regression has been demonstrated in animal models. These studies show that blocking the PDGF–B/PDGFR-β interaction leads to lack of pericyte coverage, abnormal/leaking blood vessels, inhibition of NV capillary maturation and neonatal lethality.9 Specifically, animal models of corneal and choroidal neovascularization have demonstrated that administration of agents that block the PDGF-B/PDGFR-β interaction leads to pericyte stripping from the pathological neovasculature.10 Co-administration of anti-VEGF and anti-PDGF agents leads to greater NV inhibition than either agent alone. More importantly, such combination induces NV regression as well.
Established, mature, non-pathological vascularization is unaffected by inhibitors of the PDGF-B/PDGFR-β interaction. A possible hypothesis for the decreased PDGF dependency on pericytes residing on mature host vessels as opposed to proliferating NV tissue may be related to lack of PDGFR-β expression in mature pericytes. Alternatively, the nature of pericyte-EC association differs in mature and pathologically proliferating vessels. In the mature host vascular bed (non-pathologic vascular tissue), Sphingosine-1-phosphate (SP1) mediated N-Cadherin and Gap junction activated TGF-β1 leads to a stabilized pericyte-EC interaction and mural cell differentiation.11 In addition, the pericytes and EC share a common basement membrane with individual pericytes contacting multiple endothelial cells. Conversely, pericytes involved in pathological neovascularization appear to have an abnormal morphology with a looser association to the underlying EC vasculature.12
New PDGF Antagonist
A PDGF antagonist, E10030, with high specificity and affinity in
 the form of a pegylated oligonucleotide (aptamer) has been developed by Ophthotech Corp.13 E10030 is 32-mer DNA-based oligonucleotide containing 2’-methoxy and 2’-fluoro modified bases and a 40-kDa, branched PEG attached to the 5’end. E10030 binds to PDGF with a Kd of ~300 pM and blocks its binding to PDGFR-β. In vitro/in vivo studies using this compound in combination with an anti-VEGF agent have demonstrated its effectiveness at preventing the formation of NV vessels, as well as stripping of pericytes leading to regression of established NV vessels.8
the form of a pegylated oligonucleotide (aptamer) has been developed by Ophthotech Corp.13 E10030 is 32-mer DNA-based oligonucleotide containing 2’-methoxy and 2’-fluoro modified bases and a 40-kDa, branched PEG attached to the 5’end. E10030 binds to PDGF with a Kd of ~300 pM and blocks its binding to PDGFR-β. In vitro/in vivo studies using this compound in combination with an anti-VEGF agent have demonstrated its effectiveness at preventing the formation of NV vessels, as well as stripping of pericytes leading to regression of established NV vessels.8
A Phase-1 clinical study to investigate the safety and tolerability of intravitreous injections of E10030 in combination with ranibizumab has been completed. This was a first-in-human clinical trial of intravitreous injections of E10030 in a dose-escalation (0.03 mg, 0.3 mg, 1.5 mg and 3.0 mg) scheme. Treatment-naïve patients with predominantly classic and predominantly occult subtypes were enrolled with best corrected visual acuity of 20/63 to 20/200. Mean baseline ETDRS letters read was 49.1.
All dose levels investigated were well-tolerated. There were no dose-limiting toxicities at any dose level during the study and no particular safety concerns were identified.
 |
Optical coherence tomography assessment of center point and central subfield thickness showed a decrease at all time points for all treatment groups. At week 12 there was a decrease in center point thickness of 38.9 percent overall and for central subfield thickness, a mean decrease of 33.7 percent.
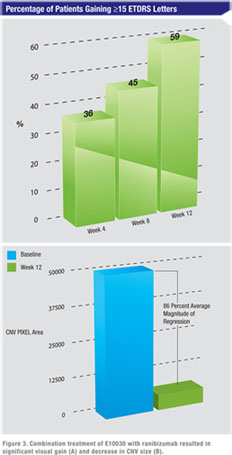
Fluorescein angiography analysis demonstrated neovascular regression (decrease in CNV size from baseline) in all patients from baseline to week 12 as read by independent readers. The vast majority had a dramatic reduction of CNV size. The mean decrease in magnitude of CNV size was 85.5 percent from baseline.
In conclusion, E10030 was well-tolerated when administered in combination with ranibizumab. There were no safety concerns identified. Although this was a phase 1 safety study with no control group, the marked enhanced vision (59 percent significant visual gain versus ~33 percent in historical controls) coupled with unprecedented level of NV regression is consistent with preliminary indication of potential efficacy.
This was the first human trial of an anti-PDGF agent administered in combination with an anti-VEGF agent in patients with wet AMD. Beyond the excellent safety profile observed for this combination regimen, initial results suggest a synergistic effect of the two mechanisms in agreement with the in vitro and in vivo studies of neovascularization. A highly powered (445 patients) Phase II trial is currently in progress. This is a randomized, controlled, six-month trial designed to compare the efficacy of the combination of E10030 plus an anti-VEGF agent (ranibizumab) versus monotherapy anti-VEGF agent (ranibizumab) in patients with wet AMD. The primary endpoint is significant visual gain (proportion of patients whose visual acuity improved from baseline by 15 or more ETDRS letters) at three months.
Dr. Mones is a vitreoretinal specialist. He is the director of the Institut de la Màcula i de la Retina at the Centro Médico Teknon. Contact him at Vilana 12, 08022 Barcelona, Spain, e-mail: assistant@institutmacularetina.com.
1. Brown DM, Kaiser PK, Michels M, Soubrane G, et al; ANCHOR Study Group. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med 2006; 355(14):1432-1444.
2. Rosenfeld PJ, Brown DM, Heier JS, Boyer DS, et al; MARINA study group. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med 2006; 355(14):1419-1431.
3. Regillo CD, Brown DM, Abraham P, Yue H, et al. Randomized, double-masked, sham-controlled trial of ranibizumab for neovascular age- related macular degeneration: PIER study year 1. Am J Ophthalmol 2008;145:239-248.
4. Zebrowski BK, Yano S, Liu W, Shaheen RM, et al. Vascular endothelial growth factor levels and induction of permeability in malignant pleural effusions. Clin Cancer Res 1999;5:3364-3368.
5. Reinmuth N, Liu W, Jung YD, Ahmad SA, Shaheen RM, Fan F, Bucana CD, McMahon G, Gallick GE, Ellis LM. Induction of VEGF in perivascular cells defines a potential paracrine mechanism for endothelial cell survival. FASEB J. 2001 May;15(7):1239-41.
6. Armulik A, Abramsson A, Betsholtz C. Endothelial/Pericyte Interactions. Circ Res 2005; 97:512-523.
7. Benjamin LM, Hemo I, Keshet E. A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Development 1998;125:1591-1598.
8. Mitchell TS, Bradley J, Robinson GS, Shima DT, Ng Y-S. RGS5 expression is a quantitative measure of pericyte coverage of blood vessels. Angiogenesis 2008;11:141-151.
9. Hall AP. Review of the pericyte during angiogenesis and its role in cancer and diabetic retinopathy. Toxicol Pathol 2006; 34:763–775.
10. Jo N, Mailhos C, Ju M, Cheung E, Bradley J, Nishijima K, Robinson GS, Adamis AP, Shima DT. Inhibition of platelet-derived growth factor B signaling enhances the efficacy of anti-vascular endothelial growth factor therapy in multiple models of ocular neovascularization. Am J Pathol 2006;168 (6):2036-2053.
11. Gaengel K, Genove´ G, Armulik A, Betsholtz C. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol 2009;29:630-638.
12. Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RK, McDonald DM; Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol 2002;160(3):985-1000.
13. Pendergrast PS, Marsh HN, Grate D, Healy JM, Stanton M; Nucleic acid aptamers for target validation and therapeutic applications. J Biomol Tech 2005;16:224–234.
Fluorescein angiography analysis demonstrated neovascular regression (decrease in CNV size from baseline) in all patients from baseline to week 12 as read by independent readers. The vast majority had a dramatic reduction of CNV size. The mean decrease in magnitude of CNV size was 85.5 percent from baseline.
In conclusion, E10030 was well-tolerated when administered in combination with ranibizumab. There were no safety concerns identified. Although this was a phase 1 safety study with no control group, the marked enhanced vision (59 percent significant visual gain versus ~33 percent in historical controls) coupled with unprecedented level of NV regression is consistent with preliminary indication of potential efficacy.
This was the first human trial of an anti-PDGF agent administered in combination with an anti-VEGF agent in patients with wet AMD. Beyond the excellent safety profile observed for this combination regimen, initial results suggest a synergistic effect of the two mechanisms in agreement with the in vitro and in vivo studies of neovascularization. A highly powered (445 patients) Phase II trial is currently in progress. This is a randomized, controlled, six-month trial designed to compare the efficacy of the combination of E10030 plus an anti-VEGF agent (ranibizumab) versus monotherapy anti-VEGF agent (ranibizumab) in patients with wet AMD. The primary endpoint is significant visual gain (proportion of patients whose visual acuity improved from baseline by 15 or more ETDRS letters) at three months.
Dr. Mones is a vitreoretinal specialist. He is the director of the Institut de la Màcula i de la Retina at the Centro Médico Teknon. Contact him at Vilana 12, 08022 Barcelona, Spain, e-mail: assistant@institutmacularetina.com.
1. Brown DM, Kaiser PK, Michels M, Soubrane G, et al; ANCHOR Study Group. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med 2006; 355(14):1432-1444.
2. Rosenfeld PJ, Brown DM, Heier JS, Boyer DS, et al; MARINA study group. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med 2006; 355(14):1419-1431.
3. Regillo CD, Brown DM, Abraham P, Yue H, et al. Randomized, double-masked, sham-controlled trial of ranibizumab for neovascular age- related macular degeneration: PIER study year 1. Am J Ophthalmol 2008;145:239-248.
4. Zebrowski BK, Yano S, Liu W, Shaheen RM, et al. Vascular endothelial growth factor levels and induction of permeability in malignant pleural effusions. Clin Cancer Res 1999;5:3364-3368.
5. Reinmuth N, Liu W, Jung YD, Ahmad SA, Shaheen RM, Fan F, Bucana CD, McMahon G, Gallick GE, Ellis LM. Induction of VEGF in perivascular cells defines a potential paracrine mechanism for endothelial cell survival. FASEB J. 2001 May;15(7):1239-41.
6. Armulik A, Abramsson A, Betsholtz C. Endothelial/Pericyte Interactions. Circ Res 2005; 97:512-523.
7. Benjamin LM, Hemo I, Keshet E. A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Development 1998;125:1591-1598.
8. Mitchell TS, Bradley J, Robinson GS, Shima DT, Ng Y-S. RGS5 expression is a quantitative measure of pericyte coverage of blood vessels. Angiogenesis 2008;11:141-151.
9. Hall AP. Review of the pericyte during angiogenesis and its role in cancer and diabetic retinopathy. Toxicol Pathol 2006; 34:763–775.
10. Jo N, Mailhos C, Ju M, Cheung E, Bradley J, Nishijima K, Robinson GS, Adamis AP, Shima DT. Inhibition of platelet-derived growth factor B signaling enhances the efficacy of anti-vascular endothelial growth factor therapy in multiple models of ocular neovascularization. Am J Pathol 2006;168 (6):2036-2053.
11. Gaengel K, Genove´ G, Armulik A, Betsholtz C. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol 2009;29:630-638.
12. Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RK, McDonald DM; Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol 2002;160(3):985-1000.
13. Pendergrast PS, Marsh HN, Grate D, Healy JM, Stanton M; Nucleic acid aptamers for target validation and therapeutic applications. J Biomol Tech 2005;16:224–234.


