Workup, Diagnosis and Treatment
 |
The initial differential included benign and malignant neoplasms, inflammatory and autoimmune diseases, chronic infection and vascular malformations. Primary malignancies of the sinus, orbit and lacrimal sac were of greatest concern upon presentation. Manifestations of diffuse large B-cell lymphoma and mantle cell lymphoma were felt to be less likely given her absence of systemic “B symptoms.” Inflammatory and autoimmune disorders such as sarcoidosis, IgG4-related disease, granulomatosis with polyangiitis (Wegener’s granulomatosis), eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) and histiocytosis were also given serious consideration. Acute bacterial infections were deemed less likely, although sino-orbital aspergillosis remained on the differential given its potential to mimic noninfectious orbital inflammation.
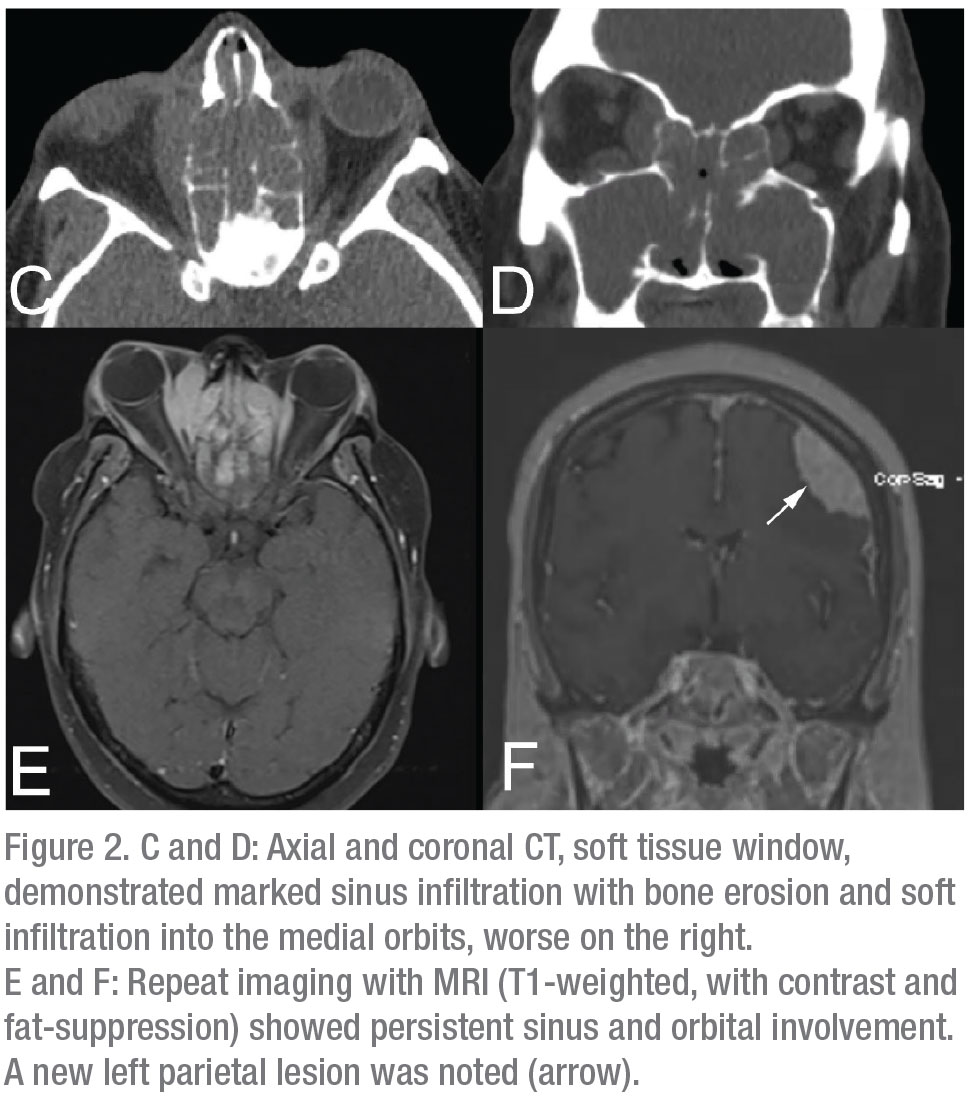
A CT scan of the head, orbits and maxillofacial bones, without contrast, was obtained as the first step. It showed a large soft tissue mass filling the frontal, ethmoid and maxillary sinuses bilaterally, with extension into the nasal cavities and orbits (Figure 2 C,D). An extra-axial mass along the left frontal lobe with parenchymal edema was also noted, along with bilateral cervical lymphadenopathy. Initial lab studies showed a normal CBC, BMP and LFTs. ESR was found to be elevated to 97 (normal: 0 to 40). Autoimmune serologies such as ANA, ANCA panel, RF, ACE and serum IgE levels were within normal limits. Serum IgG was elevated to 2,420 (normal: 700 to 1,600) and IgG4-specific levels were elevated to 159 (normal: 2 to 96). An MRI of the brain, orbits, and sinuses was obtained with contrast, to better delineate the extent of the soft tissue mass. This confirmed CNS involvement near the left frontal lobe (Figure 2 E,F). A transnasal endoscopic biopsy of the sinonasal mass was then performed in order to obtain a tissue diagnosis.
 |
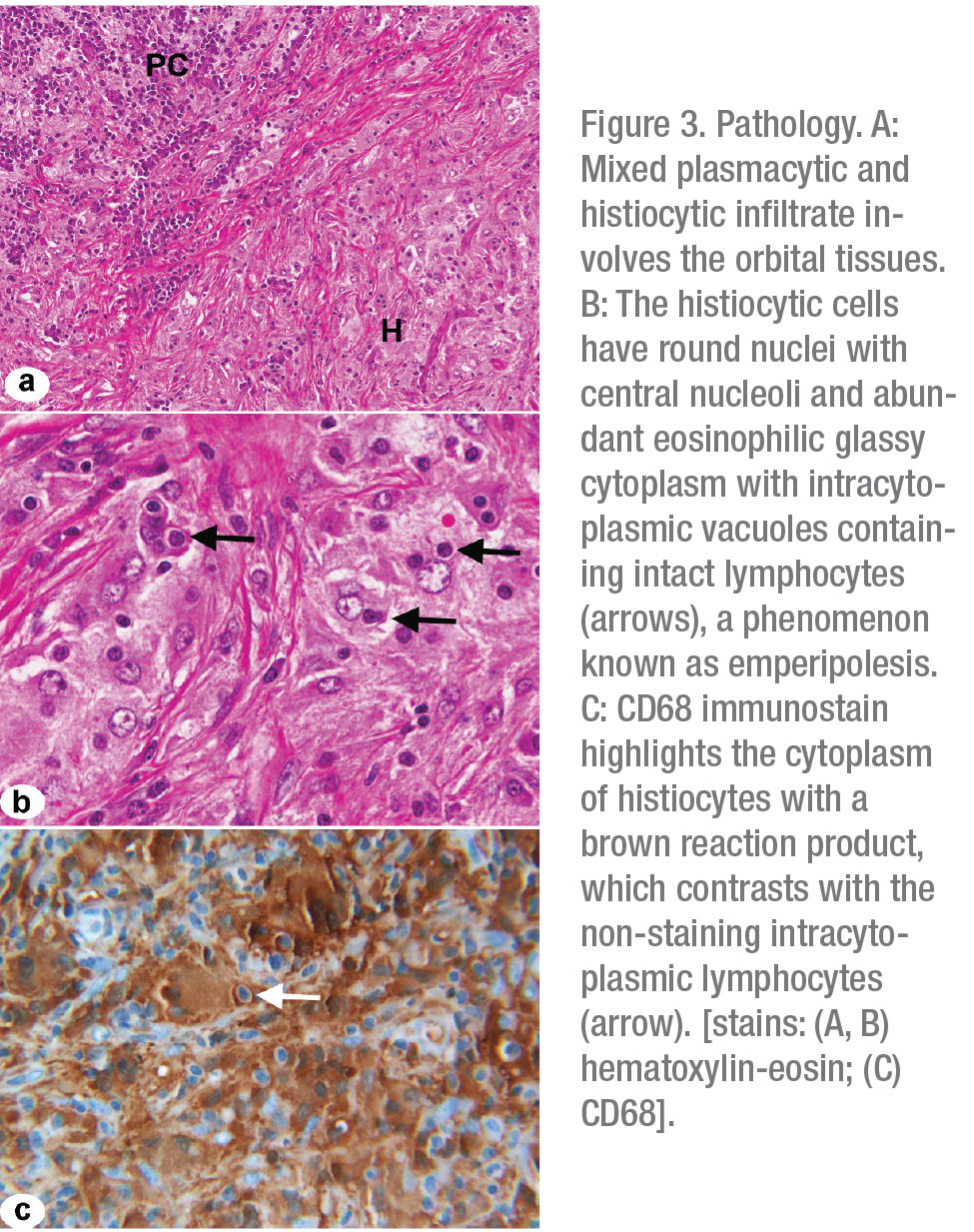
Final pathology from the nasal mass revealed fibrosis with a polymorphous infiltrate containing histiocytes and large macrophages with intracytoplasmic lymphocytes, consistent with a diagnosis of Rosai-Dorfman Disease (Figure 3 A-C). The patient was started on an extended course of oral prednisone and underwent neurosurgical debulking of her left frontoparietal mass. Her prednisone was tapered and she was transitioned to sirolimus after her procedure. Several months later, she was hospitalized for bilateral pneumonia, and her sirolimus was discontinued, given concern that her infection was triggered by sirolimus-induced pneumonitis.
Without receiving any systemic treatment, over the next several months she reported worsening headaches and sinus pressure, and repeat MRI showed enlargement of the paranasal sinus mass with further infiltration of the right orbit. Recurrence of the frontal lobe mass was also noted on imaging, with an additional focus of axial enhancement along the anterolateral right frontal lobe. At the time of this report, the patient had restarted systemic treatment with methotrexate and 6-mercaptopurine and is scheduled to follow up with pulmonology, oncology, neurosurgery and ENT over the next several months for ongoing management.
Discussion
Rosai-Dorfman Disease, also known as sinus histiocytosis with massive lymphadenopathy, is a non-malignant proliferation of histiocytes in lymph node sinuses and many other extranodal sites.1 Clinical manifestations of RDD may include classic B symptoms of bilateral painless cervical lymphadenopathy, intermittent fevers, night sweats and weight loss.2 Extranodal RDD occurs in approximately 43 percent of cases; it may include cutaneous, CNS, intrathoracic, retroperitoneal and osseous manifestations. Ophthalmic involvement occurs in 10 percent of cases, presenting as a mass in the orbital soft tissues, eyelids, lacrimal glands or other locations.3 Orbital RDD has rarely been reported in isolation; it generally occurs unilaterally. CNS involvement is even rarer, occurring in less than 5 percent of cases in the literature, although the risk is greater in elderly patients.2-4
RDD is considered a self-limited disorder of unknown etiology, although recent studies have suggested associations with mutations in NRAS, KRAS and ARAF. These genes are involved in cell growth and development, and mutations are believed to be closely linked to tumorigenesis.5 Diagnosis and staging of patients with RDD should include comprehensive imaging of the neck, chest, abdomen and pelvis, with consideration for PET/CT during the initial survey.1,2,4 Patients with orbital or neurologic involvement, as described in our own case, should undergo MRI of the brain, orbits and spine, with gadolinium.6 Although serum studies may offer some value in ruling out alternative diagnoses, definitive diagnosis is made only through tissue biopsy.2,7 Pathologically, RDD is typified by histiocytic proliferation with infiltration of mature plasma cells and lymphocytes. A unique histologic feature of this disease is the finding of emperipolesis, whereby histiocytes phagocytize viable lymphocytes without enzymatic degradation.2
No uniform approach to treatment has been established for RDD, and treatment must be tailored to each patient. Initial observation is prudent in patients with only nodal or cutaneous disease, as between 20 to 50 percent of cases undergo spontaneous remission.5 In cases with symptomatic CNS involvement or organ dysfunction secondary to compression, surgical debulking may achieve symptomatic control and restore function. Surgical excision is considered the first-line therapy for orbital masses, which tend to be more progressive and symptomatic than lesions at other sites.1,2 Those with disseminated or multifocal disease not amenable to surgical resection are often treated with intralesional or systemic steroids, which may reduce nodal size and symptoms. Relapses are common, and durable responses to steroids alone have not been consistently achieved in the literature.2,4,5,8
Chemotherapy for treatment of vision-threatening compressive optic neuropathy in combination with primary surgical excision has been advocated by many authors. Combination therapy with CHOP-like regimens or methotrexate and 6-mercaptopurine have demonstrated some promise in the recent literature, although many other agents are under investigation. Low-dose salvage radiotherapy after surgical debulking has also been proposed, but guidelines for standard dosing haven’t been established.1,2,4 RDD has a variable clinical course, and several disease patterns including stable, recurrent or progressive disease have been described. Mortality from RDD approaches 7 percent and is primarily a consequence of direct renal or CNS invasion. Careful follow-up is necessary to monitor for malignant transformation and additional systemic complications.2
In conclusion, Rosai-Dorfman Disease is a rare disorder which presents significant diagnostic and therapeutic challenges. A high index of suspicion may facilitate diagnosis, but even early recognition and treatment may not alter the clinical course of the disease. Although classified as benign, Rosai-Dorfman Disease can have severe systemic complications, and orbital involvement necessitates timely and aggressive treatment in order to preserve a patient’s sight. REVIEW
1. Abla O, Jacobsen E, Picarsic J, et al. The spectrum of orbital Rosai-Dorfman disease. Ophthal Plast Reconstr Surg 2019;32:2:56-59.
2. McClellan SF, Ainbinder DJ. Orbital Rosai-Dorfman disease: A literature review. Orbit 2013;32:5:341-346.
3. Su X, Zhang L. Orbital Rosai–Dorfman disease: A case report and literature review. J Int Med Res 2019;47:11:5891-5895.
4. Tan JJ, Narang S, Purewal B, et al. Extranodal Rosai-Dorfman disease of the orbit: Clinical features of 8 cases. Ophthal Plast Reconstr Surg 2016;32:6:458-461.
5. Abla O, Jacobsen E, Picarsic J, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood 2018;131:26:2877-2890.
6. Mohadjer Y, Holds JB, Rootman J, Wilson MW, Gigantelli JW, Custer PL. The spectrum of orbital Rosai-Dorfman disease. Ophthal Plast Reconstr Surg 2006;22:3:163-168.
7. Wen JH, Wang C, Jin YY, et al. Radiological and clinical findings of isolated meningeal Rosai-Dorfman disease of the central nervous system. Medicine (Baltimore) 2019;98:19:e15365.
8. Skoog L, Tani E. Histiocytic and dendritic neoplasms. Monogr Clin Cytol 2009;18:56-59.


