Workup, Diagnosis and Treatment
 |
The differential diagnosis of progressive, dome-shaped, yellow-orange subepithelial epibulbar nodule in a young patient includes reactive and inflammatory lesions, such as pyogenic granuloma, juvenile xanthogranuloma, nodular fasciitis and keloid. Benign neoplasms, such as dermolipoma/lipoma, amelanotic conjunctival nevus, fibrous histiocytoma, myxoma and conjunctival stromal tumor/ fibroma are also diagnostic considerations. Amyloid deposits and lymphoproliferative lesions, including benign reactive lymphoid hyperplasia and pediatric follicular lymphoma, may present similarly. Other malignant neoplasms, such as melanoma and various sarcomas are much less likely given the patient’s age and the overall benign appearance of the lesion.
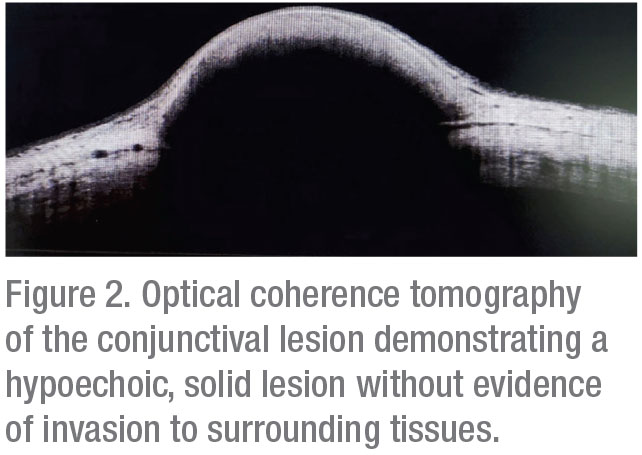
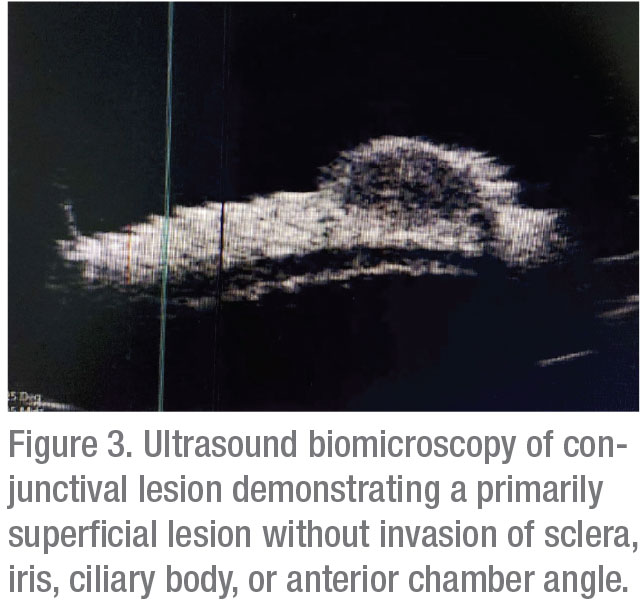
Corneal topography was subsequently obtained, which documented regular astigmatism; no other corneal pathology was noted. Optical coherence tomography demonstrated a well-circumscribed hypoechoic, solid lesion (Figure 2). Ultrasound biomicroscopy showed a hypoechoic, primarily superficial lesion without significant invasion of the sclera, anterior chamber angle, ciliary body or iris (Figure 3).
 |
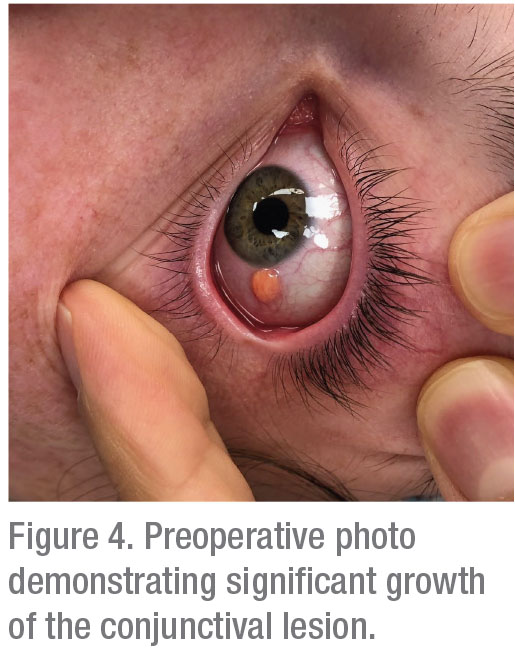
Following imaging modalities, treatment options were discussed, including observation, topical corticosteroids and excisional biopsy. Ultimately, because neoplastic diseases were considered, the excisional biopsy with double freeze-thaw cryotherapy and conjunctivoplasty were performed. On the operative day, which was approximately six weeks after the patient’s initial presentation, the lesion was noted to have significant growth (Figure 4).
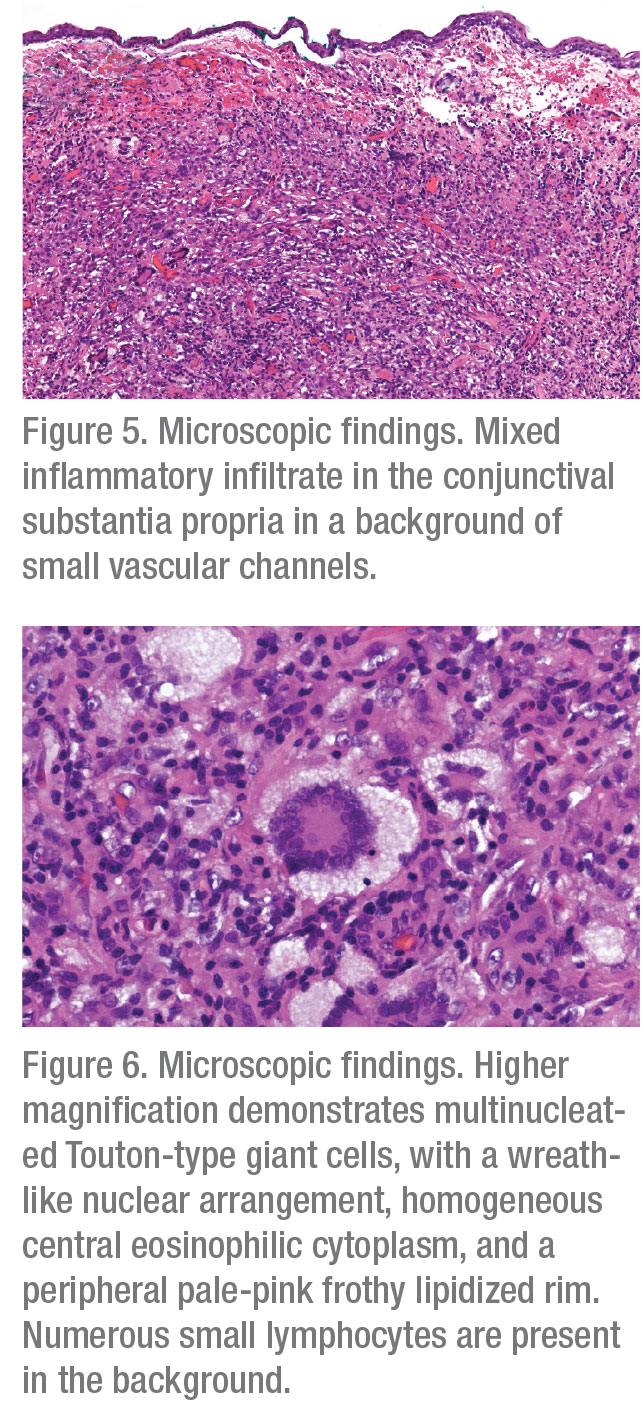
Histopathologic analysis of the lesion demonstrated a stromal infiltrate of macrophages, including xanthoma cells and Touton-type giant cells, lymphocytes and rare eosinophils in a background of small vascular channels (Figures 5 and 6). Immunohistochemical stains showed that macrophages stained positive for a macrophage marker CD163 and negative for Langerhans’ cell markers S100 and C1a. Although the macrophages were negative for mutant BRAF V600E, there was a strong and diffuse nuclear positivity for Cyclin D1, suggestive of mitogen-activated protein kinase (MAPK) signaling pathway activation. The combined clinical, morphologic, and immunohistochemical findings were compatible with an epibulbar (corneoscleral limbal) juvenile xanthogranuloma (JXG).
 |
The patient experienced no immediate postop complications. She was seen postoperative day one, presenting with a subconjunctival hemorrhage of the left eye. At the surgical site the limbal sutures were intact with well-apposed conjunctiva. Vision was stable. On postoperative day one she was started on a topical neo-poly-dex four-week taper. She was seen postoperative week one and showed a resolving subconjunctival hemorrhage. Sutures remained in place, and no evidence of tumor recurrence was evident. Vision remained stable. The patient was advised to continue her topical neo-poly-dex taper, and to follow-up in six weeks for further evaluation.
Discussion
 |
JXG is defined as a collection of histiocytes within a granulomatous reaction that doesn’t meet criteria for the diagnosis of Langerhans’ cell histiocytosis (LCH). JXG is the most common of non-LCH histiocytoses, classically presenting as single or multiple red-yellow-brown papules and/or nodules of the face, neck and trunk.1,2 Although up to three-quarters of JXG lesions arise within the first year of life, JXG can also present in older patients.2
While the majority of JXG lesions involve the skin, the disorder can involve extracutaneous sites, with the eye the most frequently affected.3 In a retrospective interventional case series of 30 patients with ocular JXG, the iris was the most common site (n=21, 68 percent), with the conjunctiva (n=6, 19 percent), eyelid (n=2, 6 percent), choroid (n=2, 6 percent), and orbit (n=1, 3 percent) less commonly affected.4 Presenting symptoms of ocular JXG include spontaneous hyphema (most common), increased IOP, blurry vision, heterochromia, eye redness and conjunctival mass.4 An adult variant of JXG which shares many clinical and histologic similarities, called adult-onset xanthogranuloma (AOX), typically presents as an orbital mass or a nodule at the corneoscleral limbus, which progressively increases in size.5 Pediatric JXG can be disseminated and can be associated with other systemic diseases, including neurofibromatosis 1 (NF1), Niemann-Pick disease, urticaria pigmentosa and juvenile myelomonocytic leukemia (JMML).5,6 AOX is an isolated lesion, and is confined to a singular ocular region.7 It’s important to note that periocular xanthogranulomatous orbital disease in adults can occur in a setting of adult-onset asthma, necrobiotic xanthogranuloma (associated with hematolymphoid malignancies) and Erdheim-Chester disease (a systemic histiocytic/dendritic cell neoplasm with frequent BRAF V600E mutations). Thus, the diagnosis of isolated AOX should be made only after careful exclusion of xanthogranulomatous diseases that have systemic implications.
JXG and AOX are diagnosed clinically with histopathologic confirmation. Clinical exam classically shows a well circumscribed, nodular or dome-shaped yellow-to-orange lesion. Histopathologic analysis of both entities classically shows a collection of foamy histiocytes, lymphocytes, fibroblasts, eosinophils and Touton-type giant cells. The presence of Touton-type giant cells is a characteristic finding in various xanthogranulomatous diseases, including JXG/AOX.8,9 Immunohistochemically, JXG and AOX macrophages demonstrate positivity for CD68 and CD163, and are negative for Langerhans’ cell markers for S100, CD1a, and Langerin, which distinguishes JXG/AOX from LCH.10
Many theories have been proposed regarding the pathogenesis of JXG/AOX, however at this time the exact cause is not clearly understood. One hypothesis suggests that localized JXG/AOX may originate from a histioxanthomatous reaction response to local tissue injury.11 Some investigators suggested a potential relationship between JXG/AOX and cytomegalovirus (CMV) infection; however, there is no firm evidence to substantiate such an association.12 Development of JXG following treatment for T-ALL and LCH in some patients has led to a theory proposing that chemotherapy induces transformation of Langerhans’ cells and clonally similar T-lymphoblast leukemic/lymphoblastic precursors into foamy cells.13,14 Further research has led to the theory that an LCH-induced cytokine storm leads to the development of JXG.15 Recent evidence suggests that JXG belongs to a group of “inflammatory histiocytic/ dendritic neoplasms,” derived from a histiocytic/dendritic stem cell precursor, which is mutated and clonally expanded. This theory is supported by identification of MAPK/ERK pathway mutations in JXG, although BRAF V600E mutations (common in LCH and Erdheim-Chester disease) are not typical of JXG.16,17
The treatment of JXG varies and is dependent upon location. While cutaneous JXG typically stabilizes or regresses in one to five years, treatment of periocular and ocular JXG is often required due to potential visual complications if the disease is allowed to run its natural course.17 Excisional biopsy is a frequent treatment modality for conjunctival and eyelid lesions, which has the advantage of being both diagnostic and therapeutic. Conjunctival and eyelid JXG have also shown response to topical and intralesional corticosteroids.4,18 Orbital JXG has been documented to undergo complete resolution with excisional biopsy as well as conservative observation.4,19 Choroidal JXG has been noted to resolve following systemic steroids and radiation therapy. Immediate treatment for iridic JXG is imperative, as studies have shown therapy is required to avoid complications such as iritis, severe hyphema, secondary glaucoma, neovascularization, vision loss and enucleation. High-dose topical corticosteroids with a prolonged taper over three to four months has proven effective in most of iridic JXG cases. For refractory cases, periocular corticosteroids, systemic steroids, excisional biopsy and low-dose ocular radiotherapy (in escalating order) have all shown appropriate responses.4 Surgical excision is the gold standard for management of AOX. Reportedly, AOX responds poorly to topical steroids; intralesional steroids and/or excisional biopsy are required for disease resolution.8
Overall, the prognosis for ocular JXG/AOX is excellent. A review of 30 ocular JXG patients demonstrated tumor regression in all patients following treatment, with only two patients experiencing persistent neovascularization of the iris and elevated IOP.4 None of the eyes required enucleation. In one review of AOX, all patients (n=10) showed complete resolution without recurrence following excisional biopsy.8
In conclusion, JXG/AOX should be considered in a patient with an elevated dome-shaped yellow-orange epibulbar nodule adjacent to the corneoscleral limbus. Clinical-pathologic correlation is required to exclude the potential systemic associations. While the exact etiology of JXG remains unclear, the recent evidence suggests that JXG belongs to a spectrum of inflammatory histiocytic-dendritic neoplasms. Topical/intralesional steroids and excisional biopsy remain the mainstay of treatment for ocular JXG/AOX, with systemic corticosteroids and ocular radiotherapy being reserved for recalcitrant cases. While most patients experience complete tumor regression without significant visual sequelae following therapy, it’s important to monitor patients for recurrence and associated symptoms such as glaucoma, neovascularization and vision loss. REVIEW
1. Cypel TKS, Zuker RM. Juvenile xanthogranuloma: Case report and review of the literature. The Canadian Journal of Plastic Surgery 2008;16:3:175–177.
2. Szczerkowska-Dobosz A, Kozicka D, Purzycka-Bohdan D, Biernat W, Stawczyk M, Nowicki R. Juvenile xanthogranuloma: A rare benign histiocytic disorder. Postepy Dermatol Alergol 2014;31:3:197-200.
3. Freyer DR, Kennedy R, Bostrom BC, et al. Juvenile xanthogranuloma: Forms of systemic disease and their clinical implications. J Pediatr 1996;129:227–37.
4. Samara WA, Khoo CT, Say EA, et al. Juvenile xanthogranuloma involving the eye and ocular adnexa: Tumor control, visual outcomes, and globe salvage in 30 patients. Ophthalmology 2015;122:10:2130-2138.
5. Sivak-Callcott JA, Rootman J, Rasmussen SL, Nugent RA, White VA, Paridaens D, et al. Adult xanthogranulomatous disease of the orbit and ocular adnexa: New immunohistochemical findings and clinical review. Br J Ophthalmol 2006;90:5:602–608.
6. Zvulunov A, Barak Y, Metzker A. Juvenile xanthogranuloma, neurofibromatosis, and juvenile chronic myelogenous leukemia. World statistical analysis. Arch Dermatol 1995;131:8:904-8.
7. Mohamed SR, Matthews N, Calcagni A. Juvenile xanthogranuloma of the limbus in an adult. Arch Ophthalmol 2002;120:7:976–977.
8. Kontos G, Borooah S, Khan A, Fleck BW, Coupland SE. The epidemiology, clinical characteristics, histopathology and management of juvenile- and adult-onset corneoscleral limbus xanthogranuloma. Graefes Arch Clin Exp Ophthalmol 2016;254:3:413-420.
9. Hermel M, Donner A, Remky A. New treatment option for adult-onset limbal xanthogranuloma. Cornea 2010;29:1:113–116.
10. Dehner LP. Juvenile xanthogranulomas in the first two decades of life: A clinicopathologic study of 174 cases with cutaneous and extracutaneous manifestations. Am J Surg Pathol 2003;27:5:579–593.
11. Mori H, Nakamichi Y, Takahashi K. Multiple juvenile xanthogranuloma of the eyelids. Ocul Oncol Pathol 2018;4:2:73-8.
12. Vasconcelos FO, Oliveira LA, Naves MD, Castro WH, Gomez RS. Juvenile xanthogranuloma: Case report with immunohistochemical identification of early and late cytomegalovirus antigens. J Oral Sci 2001;43:1:21–25.
13. Picarsic J, Pysher T, Zhou H, et al. BRAF V600E mutation in juvenile xanthogranuloma family neoplasms of the central nervous system (CNS-JXG): A revised diagnostic algorithm to include pediatric Erdheim-Chester disease. Acta Neuropathologica Communications 2019;7:1:168.
14. Perez-Becker R, Szczepanowski M, Leuschner I, et al., An aggressive systemic juvenile xanthogranuloma clonally related to a preceding T-cell acute lymphoblastic leukemia. Pediatr Blood Cancer 2011;56:859-862.
15. Bellinato F, Maurelli M, Colato C, et al. BRAF V600E expression in juvenile xanthogranuloma occurring after Langerhans’ cell histiocytosis. Br J Dermatol 2019;180:4:933-4.
16. Chakraborty R, Hampton OA, Abhyankar H, et al. Activating MAPK1 (ERK2) mutation in an aggressive case of disseminated juvenile xanthogranuloma. Oncotarget 2017;8:28:46065–46070.
17. De Keyser C, Maudgal P, Legius E, et al. Juvenile xanthogranuloma of the corneoscleral limbus: Report of two cases. Ophthalmic Genet 2010;32;1:54–56.
18. Kuruvilla R, Escaravage Jr GK, Finn AJ, et al. Infiltrative subcutaneous juvenile xanthogranuloma of the eyelid in a neonate. Ophthalmic Plast Reconstr Surg 2009;25:4:330-2.
19. Johnson TE, Alabiad C, Wei L, Davis JA. Extensive juvenile xanthogranuloma involving the orbit, sinuses, brain and subtemporal fossa in a newborn. Ophthalmic Plast Reconstr Surg 2010;26:2:133-4.


