Presentation
A 7-year-old African-American boy presented with a pigmented conjunctival lesion in the left eye. The lesion had increased in size and vascularity compared to an examination six months prior.
Medical History
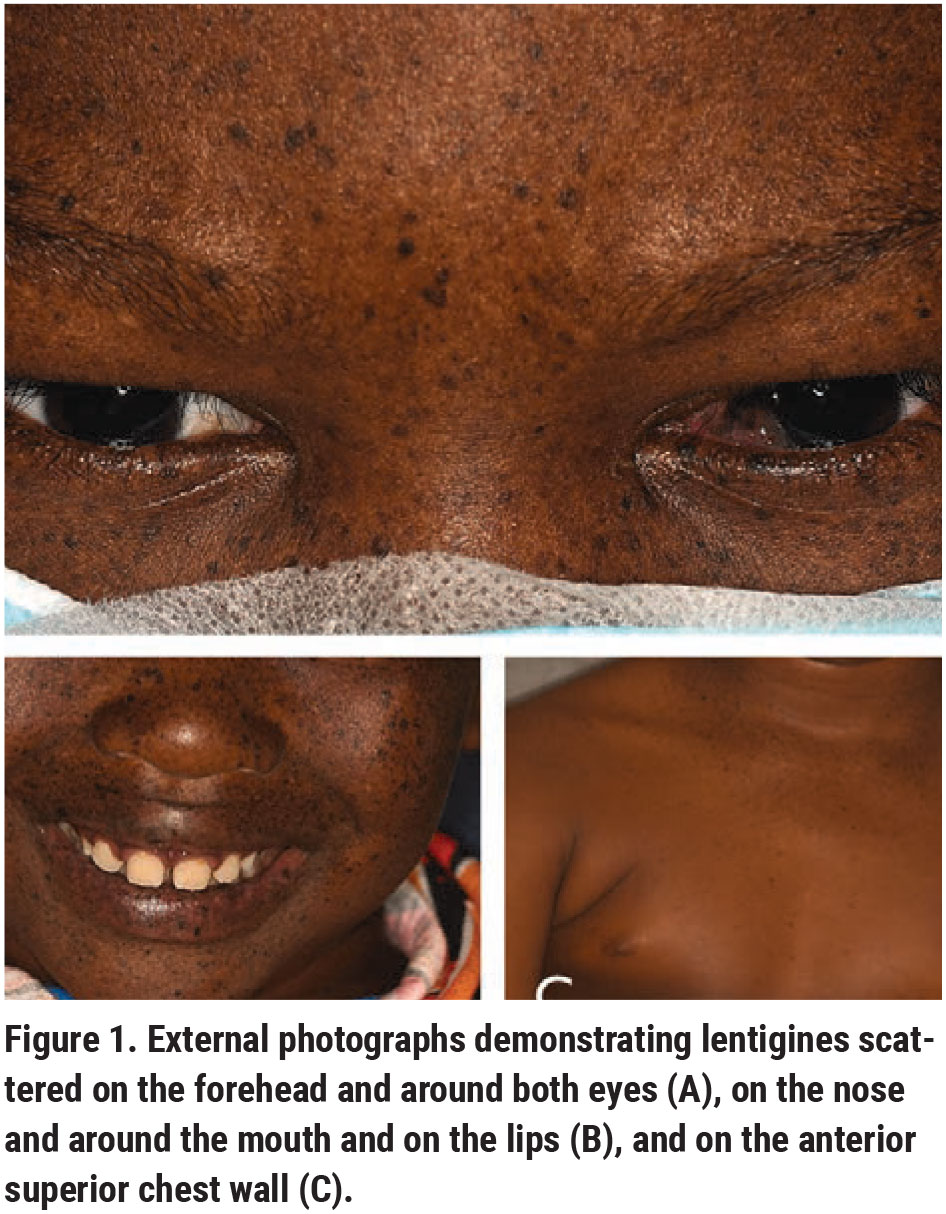 |
At 3 years of age, the patient (who was adopted, precluding the assessment of his family’s medical history) was referred to a pediatric ophthalmologist by his pediatrician for “constant squinting outdoors as long as he could remember,” and photosensitivity in both eyes. Dry skin was noted on his initial examination, and the patient’s photosensitivity was attributed to a combination of dry eye and allergic conjunctivitis. At 4 years of age, his ophthalmologist noted conjunctival pigmentation OU, as well as facial freckles. At age 5, the patient was diagnosed with a conjunctival nevus OS. At this time, the patient also underwent dermatologic evaluation for possible Peutz-Jeghers syndrome and Carney syndrome because of the numerous freckles distributed on his face, lips, torso and limbs. Genetic testing for these two conditions was ultimately negative.
Over a four-year period, the patient was managed with artificial tears, followed by ketotifen fumarate and most recently olopatadine for presumed allergic conjunctivitis. The patient continued to have persistent photosensitivity and ocular itching. During this time, he was also diagnosed with anisometropic amblyopia, myopia and astigmatism. At age 7, the conjunctival lesion OS showed growth and increased vascularity over the course of six months, prompting referral to the Ocular Oncology Service at Wills Eye Hospital. The child was otherwise healthy and meeting all developmental milestones.
Examination
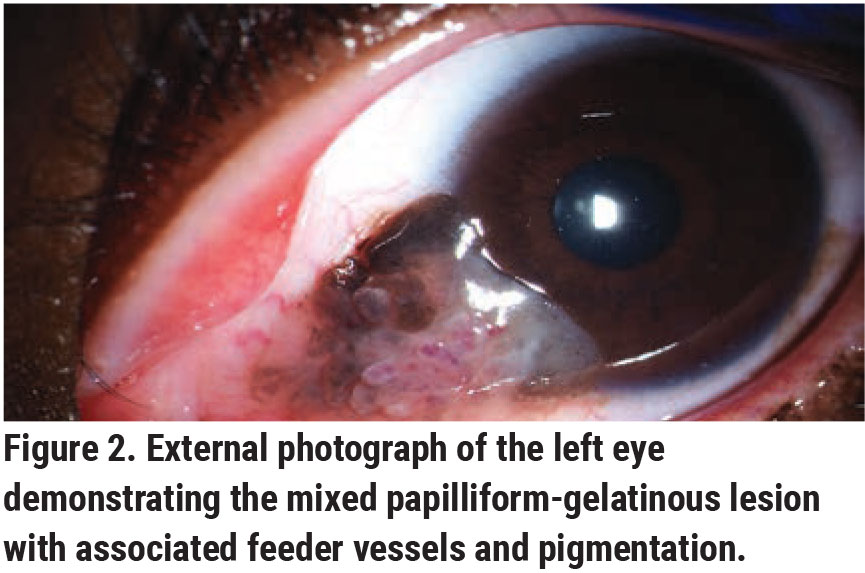 |
Ocular examination demonstrated best-corrected visual acuity of 20/50 in the right eye and 20/30 in the left. Pupils were equal and reactive, with no afferent pupillary defect. Finger tension was normal OU. Extraocular motility was full. Visual fields were full to confrontation OU. External examination showed multiple facial lentigines (Figure 1, A-C). On further physical examination, these lentigines were also noted on the buccal mucosa, chest, back, along the shins and behind the knees, but were noted to be far more concentrated in the sun-exposed regions of the skin.
Slit lamp biomicroscopy OS revealed a mixed papilliform-gelatinous lesion measuring 8 mm in basal diameter and 3 mm in thickness with associated feeder vessels and pigmentation extending across the nasal conjunctiva onto the cornea (Figure 2). Scattered complexion-associated melanosis was also present OU. The remaining ophthalmologic examination was unremarkable.
What's your diagnosis? What further workup would you pursue? The diagnosis appears below.
Diagnosis and Management
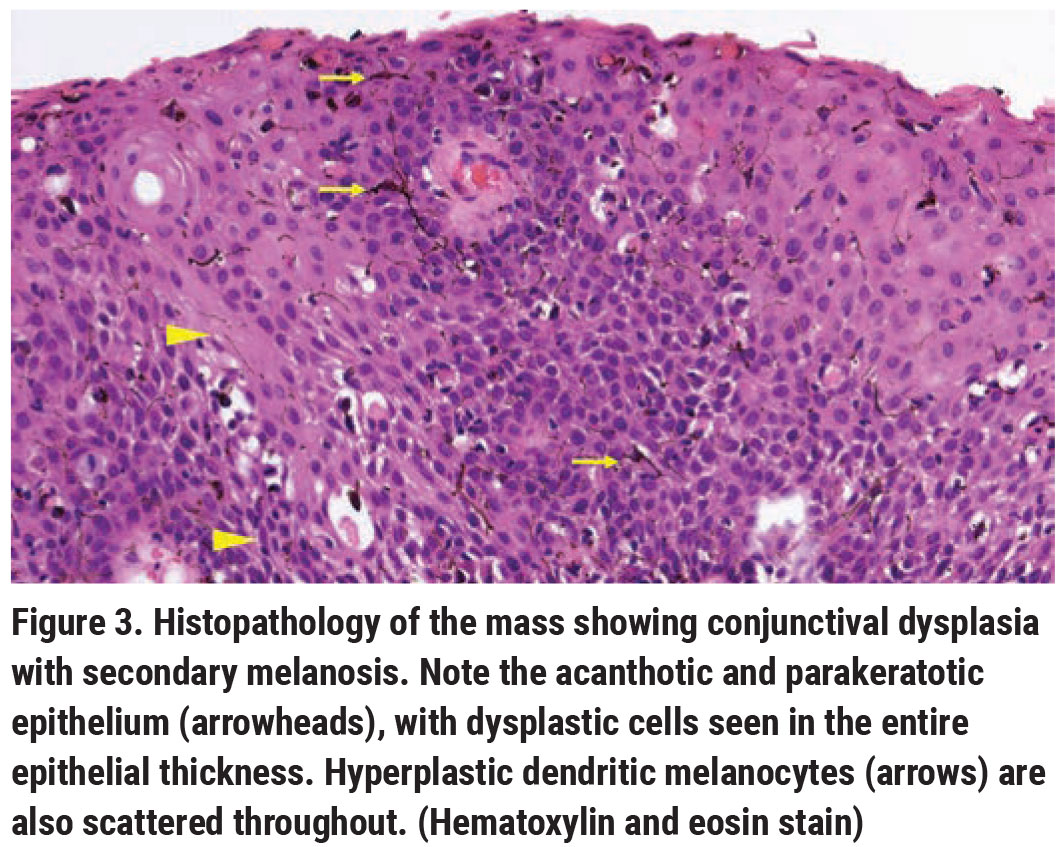 |
Given our patient’s clinical history and examination, a differential diagnosis was constructed regarding the conjunctival lesion OS specifically, as well as potential disease complexes that could be contributing to his cutaneous presentation. Benign etiologies considered included squamous papilloma, melanocytic nevus and choristomatous lesions. Although malignant conjunctival tumors are uncommon in a young child, the rapid growth and corneal involvement, irregularity and prominent vascularity raised consideration of malignant neoplasms, such as pigmented squamous cell carcinoma and conjunctival melanoma. Other considerations included inflammatory and histiocytic-dendritic disorders, such as juvenile xanthogranuloma; or a reaction to an embedded foreign body.
Excisional biopsy of the conjunctival lesion with the “no-touch technique” was performed. All margins were treated with cryotherapy, and alcohol keratectomy was performed to eliminate tumor cells from the corneal surface. The patient was placed on neomycin/polymyxin B/dexamethasone (Maxitrol) ointment for three weeks. No intraoperative or postoperative complications were encountered in the first four months of follow-up.
Pathology revealed papillomatous-like, acanthotic, parakeratotic, dysplastic epithelium, with dysplastic cells focally replacing the entire epithelial thickness (Figure 3). Foci of hemorrhage and fibrin, as well as hyperplastic dendritic melanocytes, were noted in the dysplastic epithelium. The epithelial basement membrane remained intact. These findings were compatible with conjunctival squamous cell carcinoma in situ with secondary melanosis, also known as pigmented squamous cell carcinoma in situ.
The patient’s young age at presentation with conjunctival squamous cell carcinoma was considered to be particularly noteworthy, leading to consideration of various cancer syndromes and immune dysfunction. In light of cutaneous and buccal mucosal findings, genodermatoses, such as Xeroderma Pigmentosum (XP), Carney syndrome and Peutz-Jeghers syndrome were considered. The association of conjunctival squamous cell carcinoma with a predominant sunlight-exposed skin distribution of freckles made XP a leading diagnostic consideration. Although neither Carney syndrome or Peutz-Jeghers syndrome have been previously associated with conjunctival squamous cell carcinoma, these conditions were included in the differential diagnosis because of the numerous cutaneous freckles and buccal mucosal freckling.
Genetic testing demonstrated absence of mutations in STK11, a tumor-suppressor gene implicated in Peutz-Jeghers syndrome. Testing for PRKAR1A, which has been implicated in 60 to 80 percent of cases of Carney Syndrome, was also negative. The geneticists recommended further screening and imaging, as these negative results reportedly decrease, but do not exclude, the possibility of these two disease complexes. Genetic testing for XP is currently being pursued.
Discussion
Conjunctival ocular surface squamous neoplasia (OSSN), an umbrella term that includes conjunctival intraepithelial neoplasia (CIN), squamous cell carcinoma in situ, and invasive squamous cell carcinoma, is characterized by dysplastic cells involving the squamous epithelium of the conjunctiva. When the entire epithelium consists of dysplastic cells with an intact basement membrane, the lesion is called squamous cell carcinoma in situ.
Ocular surface squamous neoplasia can involve either the conjunctiva or the cornea, but more commonly involves both. Lesions most often arise at the nasal or temporal limbus, where they appear as gelatinous, papilliform or leukoplakic nodules, frequently associated with prominent feeder vessels. Pigmented SCC of the conjunctiva occurs infrequently.1 Pigmentation in conjunctival squamous cell carcinoma generally occurs as a result of reactive (non-neoplastic) proliferation of melanocytes within the dysplastic epithelium. This phenomenon is generally seen in patients with darker skin tone and complexion-associated melanosis.
Ocular surface squamous neoplasia classically occurs in older white males. Other risk factors include extensive sun exposure, immunosuppression (e.g., HIV), vitamin A deficiency, chronic irritants, other infections or immune-dysregulated states. Based on a series of 5,002 conjunctival tumors in patients of all ages referred to an ocular oncology tertiary care center, conjunctival tumors were found to be benign (52 percent), premalignant (18 percent), or malignant (30 percent).2 Comparatively, conjunctival tumors in children demonstrate malignancy in only 3 percent of cases.3 A diagnosis of OSSN in a child or younger individual should raise suspicion for the possibility of underlying immunodeficiency or a cancer-predisposition syndrome. One study suggested that as many as half of patients younger than 50 with OSSN have HIV.4 Patients with HIV are shown to have an increased severity of OSSN, worse prognosis and a higher chance of recurrence.5,6 Ocular surface squamous neoplasia has also been associated with leukemia and lymphoma.7
Xeroderma pigmentosum is a rare disorder of defective repeair of ultraviolet radiation-induced damage, characterized by photosensitivity with severe sunburns following minimal sun exposure, early development of lentiginous pigmentation and freckling on sun-exposed areas of the body, and—most concerning—a propensity for developing skin cancer at an early age. The median age at onset of skin basal cell carcinoma and squamous cell carcinoma in people with XP is approximately 8 years, more than 50 years earlier than in the general population.8 Patients with XP are also at an increased risk of other cutaneous and conjunctival malignancies. In a case series from India, 14 patients with XP and bilateral OSSN presented with a median age of 12 years; all patients were less than 15 years of age at presentation.9 Additionally, 43 percent of the patients presented with invasive squamous cell carcinoma, which appeared to be aggressive with a recurrence rate of 64 percent.
Commonly reported ocular symptoms in an observational case series of 89 patients with a genetically confirmed diagnosis of XP included photophobia and dry eye.10 Ophthalmic pathology manifested as ectropion, lagophthalmos, conjunctival injection, conjunctival melanosis, corneal scarring and keratopathy, pterygium and cancers of both the ocular surface and eyelids.10 The authors noted that several patients had initially presented to ophthalmologists with ocular surface signs related to their XP, before any formal diagnosis of XP had been made, and that a number of children had red, photophobic eyes, which were repeatedly diagnosed as allergic eye disease.10
The prevention of cutaneous cancers through use of sunscreen, sun-protective clothing, and UV filters on eye protective equipment such as visors or glasses is paramount in these patients. Notably, ophthalmic manifestations of XP can precede the development of the more serious components of this condition. The diagnosis of XP should be considered in younger patients presenting with ocular surface dysplasia in the presence of abnormal pigmented lesions or skin freckling.
Support provided in part by the Eye Tumor Research Foundation in Philadelphia. The funders had no role in the design and conduct of the study, in the collection, analysis and interpretation of the data, and in the preparation, review or approval of the manuscript. Carol L. Shields, MD, has had full access to all the data in the study and takes responsibility for the integrity of the data. Inquiries to: Carol L. Shields, MD, Ocular Oncology Service, 840 Walnut Street, Suite 1440, Philadelphia, PA 19107. Tel: (215) 928-3105, Fax: (215) 928-1140, Email: carolshields@gmail.com.
1. Shields CL, Manchandia A, Subbiah R, et al. Pigmented squamous cell carcinoma in situ of the conjunctiva in 5 cases. Ophthalmol 2008;115:10:1673.
2. Shields CL, Alset AE, Boal NS, et al. Conjunctival Tumors in 5002 Cases. Comparative analysis of benign versus malignant counterparts. The 2016 James D. Allen Lecture. Am J Ophthalmol 2017;173:106–133.
3. Shields CL, Sioufi K, Alset AE, et al. Clinical features differentiating benign from malignant conjunctival tumors in children. JAMA Ophthalmol 2017;135:215–24.
4. Karp CL, Scott IU, Chang TS, et al. Conjunctival intraepithelial neoplasia: A possible marker for HIV infection. Arch Ophthalmol 1996;114:257–61.
5. Gichuhi S, Macharia E, Kabiru J, et al. Risk factors for ocular surface squamous neoplasia in Kenya: A case-control study. Trop Med Int Health 2016;21:12:1522–1530.
6. Kamal S, Kaliki S, Mishra DK, et al. Ocular surface squamous neoplasia in 200 patients. Ophthalmology 2015;122:8:1688–1694.
7. Lee GA, Hirst LW. Ocular surface squamous neoplasia. Surv Ophthalmol. 1995;39:6:429–450.
8. Hebra F, Kaposi M. Xeroderma, parchment skin. On diseases of the skin including exanthemata. New Sydenham Soc 1874; 61:3:252–258.
9. Gupta N, Sachdev R, Tandon R. Ocular surface squamous neoplasia in xeroderma pigmentosum: Clinical spectrum and outcome. Graefes Arch Clin Exp Ophthalmol 2011;249:8:1217–1221.
10. Lim R, Sethi M, Morley AMS. Ophthalmic manifestations of xeroderma pigmentosum: A perspective from the United Kingdom. Ophthalmology 2017;124:1652–1661.


