Workup, Diagnosis and Treatment
 |
The patient’s clinical history and presentation were suspicious for an inflammatory or neoplastic lesion of the lacrimal system. The differential diagnosis included a host of inflammatory etiologies, including sarcoidosis, granulomatosis with polyangiitis, IgG4-related ophthalmic disease and Langerhans cell histiocytosis. Lymphoproliferation (including lymphoma), lacrimal sac carcinoma or inverted papilloma, dermatofibroma and various mesenchymal tumors were also considered. Bacterial infectious etiologies such as dacryocystitis, orbital cellulitis, orbital abscess and ethmoiditis were thought to be less likely, given the chronicity of the symptoms, though atypical infections, including fungal etiologies, were possible. Serologic testing was ordered, with resultant normal values for angiotensin converting enzyme, lactate dehydrogenase, complete blood count with differential, total immunoglobulin (Ig)G, IgG4 subclass and cytoplasmic/perinuclear/atypical perinuclear antineurophil cytoplasmic antibodies.
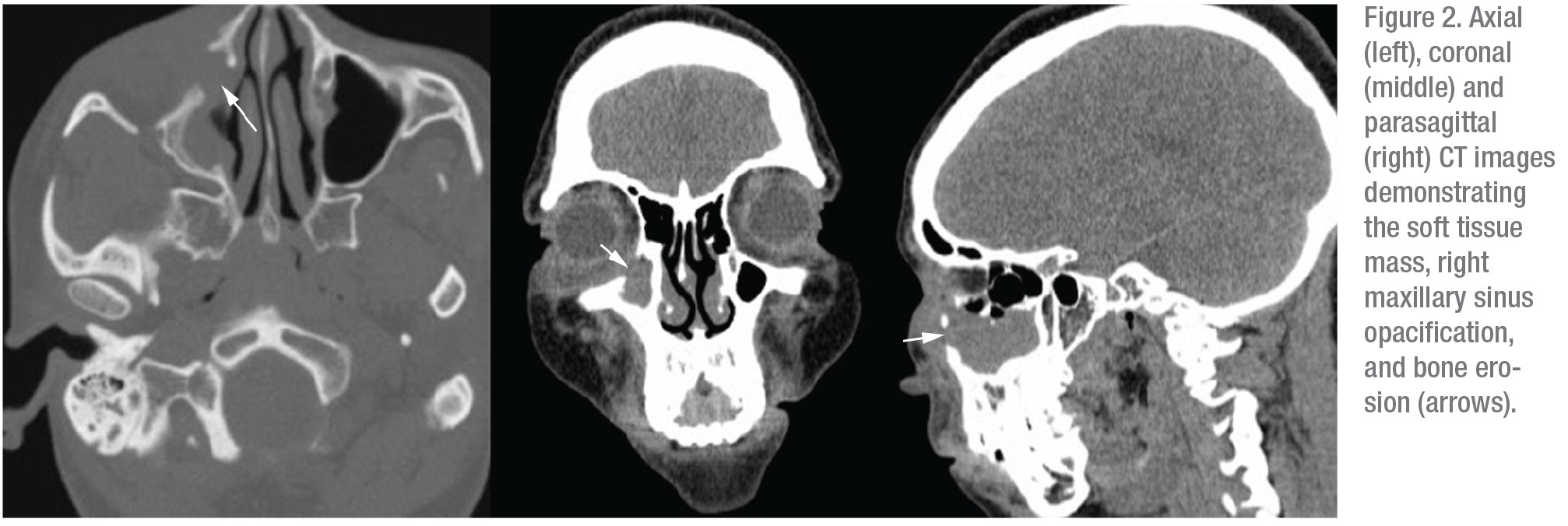
Computed tomography of the orbits and paranasal sinuses was ordered and revealed mass-like soft tissue thickening in the right medial canthus and malar soft tissue, with erosion of the right orbital floor and nasolacrimal duct, as well as the medial and anterior walls of the right maxillary sinus (Figure 2). Mild infiltration of the inferior extraconal fat and complete opacification of the right maxillary sinus were also noted.
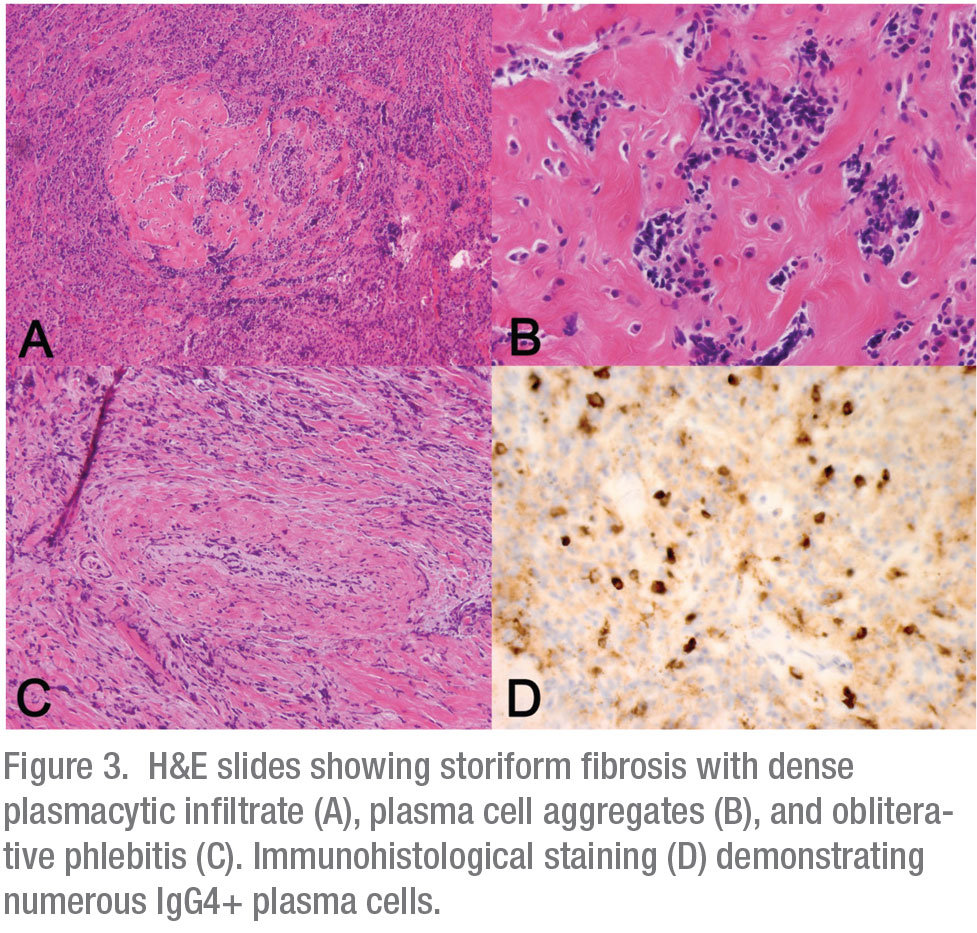
The bone eroding characteristics of this ill-defined mass were highly suspicious for a malignant process originating in the right maxillary sinus or lacrimal sac. The patient underwent a transnasal endoscopic biopsy of the lesion in the operating room. All frozen tissue specimens were consistent with an inflammatory process without evidence of neoplasia. Given the high suspicion of malignancy, a transconjunctival exploration was also performed to maximize diagnostic yield. Permanent pathologic examination of the orbital and sinus tissue revealed marked fibrosis with vaguely storiform areas, obliterative phlebitis and lymphoplasmacytic infiltrate rich in IgG4+ plasma cells (Figure 3). No malignant characteristics were present. Based on the pathologic characteristics, diagnosis of the IgG4–related disease (IgG4-RD) was made.
The patient was referred to a rheumatologist for a systemic evaluation and initiation of immunosuppressive therapy. Additional laboratory tests were performed and were found to be within the normal range, including myeloperoxidase, proteinase 3, anti-Ro/anti-La antibodies, thyroid stimulating hormone and thyroxine. The Westergren erythrocyte sedimentation rate and C-reactive protein level were elevated at 51 mm/hr and 21 mg/L, respectively. A CT of the chest, abdomen and pelvis revealed no evidence of IgG4-RD elsewhere. Before initiating immunosuppressive therapy, tuberculosis screening was performed with Quantiferon gold, with a positive result. The infectious disease consultant recommended rifampin therapy for latent tuberculosis, given the patient’s breastfeeding status. Oral prednisone was started two weeks following initiation of rifampin.
At the one month follow-up visit after initiation of corticosteroid therapy, the patient’s ocular symptoms and exam showed noticeable improvement. MRI of the orbits showed stable orbital disease. However, because of a progressive elevation in the ESR and CRP, the patient’s rheumatologist recommended a steroid taper and began rituximab infusions. Three months later, the patient reported an additional significant relief in periorbital pain. Examination demonstrated continued improvement in pericanthal edema. Mild skin lichenification was noted over her cheek, but the area was no longer tender to palpation. Repeat MRI imaging displayed no progression of her sino-orbitopathy.
 |
Discussion
IgG4-RD is an inflammatory disorder affecting one or several organ systems, characterized by significant fibrosis. It was first described in Japan in 2001 in patients with autoimmune pancreatitis (AIP) and increased serum IgG4 levels.1 It was later observed that similar lesions were found in extrapancreatic sites in many patients with AIP.2,3 Histopathologically, IgG4–RD is characterized by tumefactive lesions with obliterative phlebitis, storiform pattern fibrosis and dense lymphoplasmacytic infiltrate rich in IgG4+ plasma cells.3
Nearly one quarter of IgG4–RD cases in the United States have ophthalmic manifestations.4 When the disease affects the periocular region, it’s referred to as IgG4-related ophthalmic disease (IgG4-ROD). IgG4–ROD affects males and females equally, with mean age of onset of 55.5 Pediatric cases are rare.6
IgG4-ROD comprises 5 to 20 percent of all inflammatory orbital lesions.7 The lacrimal gland is affected in at least half of all cases, but there can also be involvement of the trigeminal nerve, extraocular muscle, orbital fat, eyelid and lacrimal drainage system.4,7 Sensory nerve infiltration is common, and bilateral infraorbital nerve involvement is a very specific feature of the disease.3 Presentation of IgG4-ROD is variable and dependent on the anatomy. Painless proptosis, restrictive strabismus with diplopia and compressive optic neuropathy have been described.8
MRI usually demonstrates a T1 isointense and T2 hypointense infiltrate that enhances with contrast administration.9 Bony destruction is rare, but possible, as demonstrated in our case.9 Of note, serum IgG4 levels are normal in 30 to 40 percent of patients.10 Moreover, increased serum IgG4 levels and presence of the IgG4+ plasma cell on immunohistochemical studies aren’t specific to IgG4-RD. Histologically, there may be an overlap between IgG4–RD and other inflammatory conditions, most notably GPA and reactive lymphoid hyperplasia.3 In addition, not all histopathologic features of IgG4-RD are necessarily present in lacrimal gland tissue.
The management of IgG4–RD requires a multidisciplinary approach. Systemic corticosteroids are considered first-line therapy, although high relapse rates are common.11 In such cases, rituximab is the agent of choice. Radiotherapy and surgical resection have met with limited success.4,12
Systemic involvement is present in over half of the IgG4–ROD cases and usually affects the salivary glands, liver, retroperitoneum, pituitary, pancreas, bile ducts and nasal mucosa.3,4 Thus, a thorough systemic evaluation is necessary and usually includes a combination of the following: measurement of pancreatic and liver enzymes; assessment of renal function; CT imaging of chest, abdomen and pelvis; and a lymph node biopsy, if lymphadenopathy is present. As reported in other chronic autoimmune conditions, IgG4–ROD increases the risk of B-cell lymphoma development, specifically extranodal marginal zone lymphoma; this transformation appears to be especially prevalent in East Asia.13
In conclusion, IgG4–ROD comprises a significant percentage of orbital inflammatory disease. It has a variable presentation and, as such, should be considered on a differential diagnosis in patients presenting with inflammatory orbital signs and symptoms. Serologic testing is neither sensitive nor specific. Biopsy is required for a definitive diagnosis. Thorough systemic evaluation is necessary in all IgG4–ROD patients, as more than half will have systemic manifestations. REVIEW
1. Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. New England Journal of Medicine 2001;344:10:732.
2. Kamisawa T, Egawa N, Nakajima H. Autoimmune pancreatitis is a systemic autoimmune disease. The American Journal of gastroenterology 2003;98:12:2811.
3. McNab A, McKelvie P. IgG4-related ophthalmic disease. Part I: background and pathology. Ophthalmic Plastic & Reconstructive Surgery 2015;31:2:83-88.
4. Wallace Z, Deshpande V, Stone J. Ophthalmic manifestations of IgG4-related disease: Single-center experience and literature review. Seminars in Arthritis and Rheumatism 2013;43:6806-817.
5. Andrew N, Kearney D, Selva D. IgG4-related orbital disease: A meta‐analysis and review. Acta Ophthalmologica 2013;91:8:694.
6. Notz G, Intili A, Bilyk JR. IgG4-related dacryoadenitis in a 13-year-old girl. Ophthalmic Plastic & Reconstructive Surgery 2014;30:6:e161-e163.
7. Plaza J, Garrity J, Dogan A, Ananthamurthy A, Witzig TE, Salomao DR. Orbital inflammation with IgG4-positive plasma cells: Manifestation of IgG4 systemic disease. Archives of Ophthalmology 2011;129:4:421-428.
8. Sane M, Chelnis J, Kozielski R, Fasiuddin A. Immunoglobulin G4–related sclerosing disease with orbital inflammation in a 12-year-old girl. Journal of American Association for Pediatric Ophthalmology and Strabismus 2013;17:5:548-550.
9. Toyoda K, Oba H, Kutomi K, Furui S, Oohara A, Mori H, Numaguchi Y. MR imaging of IgG4-related disease in the head and neck and brain. American Journal of Neuroradiology 2012;33:11:2136.
10. Sah RP, Chari ST. Serologic issues in IgG4-related systemic disease and autoimmune pancreatitis. Current Opinion in Rheumatology 2011;23:1:108-113.
11. Wu A, Andrew NH, McNab AA, Selva D. IgG4-related ophthalmic disease: pooling of published cases and literature review. Current Allergy and Asthma Reports 2015;15:6:27.
12. Kubota T, Moritani S, Katayama M, Terasaki H. Ocular adnexal IgG4-related lymphoplasmacytic infiltrative disorder. Archives of Ophthalmology 2010;128:5:577-584.
13. McNab AA, McKelvie P. IgG4-related ophthalmic disease. Part II: clinical aspects. Ophthalmic Plastic & Reconstructive Surgery 2015;31:3:167-178.


